Review Article - (2013) Volume 2, Issue 3
Alzheimer’s disease (AD) is an etiologically heterogeneous disorder. While many genes have been found to be associated with Early and Late onset AD, a large portion of the predicted heritability remains unidentified. Here we review AD pathology, with an overview of AD genetics. In addition, we review epigenetic mechanisms and the current literature that suggests a relationship between epigenetic mechanisms and AD pathology. The genome wide association studies conducted to date can explain a percentage of AD cases. The remainder may be best explained by complex interactions between epigenetic and environmental factors that differ between individuals.
Keywords: Alzheimer’s; Kinases; Epigenetics; Etiology
Alzheimer’s disease(AD) was characterized by Aloysius “Alois” Alzheimer in 1907, and was based upon his observations and treatment of a 51 year old patient named August ‘D’[1]. The patient showed symptoms of short term memory loss, unusual behavior and the neuropathological characteristics that have become the hallmarks of Alzheimer’s disease [1]. AD is the most common type of dementia, which is a term that describes a wide range of symptoms such as trouble with memory, language, ability to focus, reasoning skills, and visual perception [2]. Alzheimer’s disease is a progressive disease that is fatal given that no other cause of death intervenes. It’s also the most common form of age-related neurodegenerative dementia, a serious health problem in the industrialized world, and is currently the 6th leading cause of death in the United States [3].
The current figures from the Alzheimer’s Association states that one in three seniors will die with AD or another form of dementia. One in eight people 65 years of age and older have AD [3]. When examining people that are 85 and older, the incidence of AD increases to one in two individuals [3]. It is estimated that currently within United States, over 5 million people have AD, and this is expected to rise to over 13 million by the year 20503.Currently, it is estimated that the care provided by family, and other unpaid caregivers of people with dementia is valued at about $210 billion [4].
The two hallmarks of AD are extracellular Beta (β)-Amyloid plaques and neurofibrillary tangles [5,6]. β-Amyloid plaques are formed from cleavage of Amyloid Precursor Protein (APP), which is an integral membrane protein that is expressed throughout the body and particularly concentrated in neuronal synapses. The primary function of APP is not fully understood, but it has been implicated in neurite extension and synaptic plasticity [4]. β and Gamma (γ) secretases cleave APP to produce fragments that aggregate together to form the β-Amyloid plaques. β-secretase is an integral membrane as partyl protease encoded by the β-site APP-cleaving enzyme1 (BACE1) gene [7]. The other secretase that is involved in the production of β-Amyloid, γ-secretase, is composed of 4 subunits: presenilin 1(PSEN1), presenilin 2(PSEN2), nicrastin, and APH1 [2,4], where the active site consists of presenilin [2].
A widely held theory of AD pathogenesis is the amyloid cascade hypothesis, which states that the deposition of the β-Amyloidpeptide in the brain is the initiating event in disease pathology [8]. The amyloid hypothesis postulates that the disease is the result of an imbalance between the production and degradation of β-Amyloid [9]. Normally, β-Amyloidis degraded by peptidases such as neprilysin, insulindegrading enzyme, and endothelin-converting enzyme [2]. This central theory has strong support, from work beginning with Alois Alzheimer [1]and continuing through the deduction of the sequence of the amyloid beta protein [10] and cloning of mutations in APP [11,12], PSEN1 and PSEN2genes [13,14]. A recent development that significantly strengthened the amyloid hypothesis was the discovery by Jonssonet al. [15] of an APP mutation that reduces production of β-Amyloid and is protective against Alzheimer’s disease as well as age-related cognitive decline.It has been hypothesized that β-Amyloid protein deposition precedes neurofibrillary tangles [16], cell loss, and vascular damage [17]. In transgenic murinemodels, β-Amyloid deposition developed prior to tangles [18]. Working witha transgenic mouse model, Xu et al. [16] described an accumulation of β-Amyloid precipitateda loss of solubility of intracellular cytosolic proteins such as glycolytic enzymes and members of the chaperone family. β-Amyloidplaques have also been thought to induce neuronal oxidative stress, resulting in phospholipid peroxidation and protein oxidation in AD brain [19].
The second hallmark of AD is the presence of hyperphosphorylated microtubule associated protein tau (MAPT). The tau protein is primarily expressed in neurons [20] and has been shown to be involved with tubulin polymerization as well as acting to stabilize microtubules against depolymerization [21], stabilize microtubules responsible for axonal transport [20], increase neuritic stability, impact the rate of neurite elongation, and increase net microtubule stability [2,22]. Different iso forms of the tau protein are expressed due to alternative RNA splicing, and all iso forms are capable of forming the fibrillary tangles that are a hallmark of AD [23,24]. A balance between kinases (ex. GSK-3Beta, CDK5) and phosphatases (ex. PP-1,PP2) plays a role in regulating tau phosphorylation [2]. Tau hyper phosphorylation leads to disassembly of microtubules causing disruption in axonal transport, and impaired synaptic and neuronal function [2,25]. Hyperphosphorylated tau aggregates into filaments, causing an inability to bind and stabilize microtubules [26], and subsequent formation of neurofibrillary tangles [2,26]. In addition to the production of plaques and tangles, other pathogenic mechanisms such as oxidative stress, inflammation, cell-cycle abnormalities, and mitochondrial dysfunction [27,28] have been reported to precipitate neuropathological changes that cause degeneration of neurons and synapses in the cerebral cortex and subcortical regions of the brain [2]. Loss of neurons results in atrophy of the affected regions of the brain, including degeneration in the temporal and parietal lobes, as well as parts of the frontal cortex [29]. This neuronal atrophy has been documented by magnetic resonance imaging (MRI) and positron emission tomography (PET), as an individual progresses from mild cognitive impairment to Alzheimer’s disease [30,31]. In addition to the pathogenic mechanisms of the amyloid cascade hypothesis, AD pathology and clinical symptoms have been correlated with oxidative stress [32], inflammation [33,34], obesity [35], cardiovascular disease [36], traumatic brain injury [37-42] and diabetes [43].
Alzheimer’s disease occurs in both a familial and a sporadic form, also known as early on setor late onset Alzheimer’s disease(LOAD), respectively. Familial Alzheimer’s Disease (FAD) is an autosomal dominant disorder that is inherited in a mendelian fashion [44]. FAD accounts for a minority of the total AD cases (<5%) [45,46], and has an earlier age of onset(below 60 years of age) [47]. In comparison, LOAD has an age of onset above 65yrs [47]. FAD has been associated with mutations in the APP, Presenilin 1 (PSEN1), and Presenilin 2 (PSEN2) gene [8,24,47-49]. Mutations in the APP gene that cause FAD are clustered near the alpha- (α),β-, and γ- secretase cleavage sites, where most of these mutations increase cleavage by γ-secretase [50]. So far, 24 mutations for APP, 185 mutations for PSEN1, and 13 mutations for PSEN2 mutations have been found. All of these mutations, except one, are inherited in a mendelian autosomal dominant fashion, and are fully penetrant [47]. The early onset forms of AD fit the amyloid cascade hypothesis, where APP, PSEN1 and PSEN2 mutations increase production of β-Amyloidplaques [48,49].
Late onset Alzheimer’s disease (LOAD) results from various genetic and non-genetic factors. The strongest known genetic risk factor is carriage of the epsilon (e)4 allele at the apolipoprotein E (APOE) locus. The protein product of this gene combines with lipids to form lipoprotein molecules that are involved in packaging cholesterol and other fats as well as their transport into blood [4]. In AD pathology APOEis believed to play a role in the clearance of β-Amyloid [47]. There are three different alleles of APOE known as ε2, ε3, and ε4. These alleles code for three iso forms of the protein that differ amongst each other for amino acid residues at position 112 and 158. The ε2,ε3, and ε4 alleles code for cysteine/cysteine, cysteine/arginine, and arginine/arginine residues respectively [47]. Individuals that are homozygous for the ε4 allele are 10 to 20 times more likely to develop AD in comparison to ε4 negative individuals, and the presence of the ε2 allele in an individual has a decreased risk for AD [4]. In addition to APOE, genome wide association studies have identified eleven other loci to have an effect on LOAD. These genes are CR1,BIN1, CLU, PICALM, MS4A4/MS4A6E, CD2AP, CD33, EPHA1, SORL1, ATXN1 and ABCA7 [47,51-53]. These genetic loci have said to account for 50% of LOAD cases [52], leaving a large portion of the heritability still left unidentified. The remaining unexplained heritability within individuals that develop LOAD may be explained through a variety of epigenetic mechanisms.
The term epigenetics was coined by Conrad Waddington to describe “the branch of biology which studies the casual interactions between genes and their products, which bring the phenotype into being” [54]. Today the term broadly applies to changes in gene regulation and cellular phenotype without changes to the DNA sequence itself, as the phenotype of a cell is determined by its expression profile [55]. Epigenetic marks drive much of this expression and provide diversity to this phenotype via chromatin alteration that affects gene transcription. An epigenome is the chromatin state found across the genome at a certain time point and cell type, and therefore thousands of epigenomes can exist for a single given genome [56]. Even though there is no alteration in the DNA sequence itself, epigenetic marks, chromatin activity, and histone modifications [57] are heritable during cell division, keeping these epigenetic marks intact and passed on to dividing cells [58]. Some epigenetic modifications are stabilized and maintained throughout the life of an organism, while others change over time due to intrinsic or environmental factors [59].
A example of an epigenetic modification to DNA in mammals is the methylation of Cytosine to form 5-MethylCytosine at the C5 position in CpG dinucleotides [57,58]. DNA methylation is thought occur primarily in CpG dinucleotides and is therefore correlated to the occurrence of this DNA motif, known as CpG islands. CpG islands are short stretches of DNA where the presence of the CpG sequence is higher than in other regions which are characterized by GC rich regions of the genome [60]. These motifs are rarely larger than 5kbp and overlap with the promoter regions for 50% to 60% of human genes [60]. Most housekeeping and widely expressed genes have a CpG island covering the transcriptional start site [61], except when they are associated with imprinted genes. These islands tend to be unmethylated for housekeeping and tissue specific genes at developmental stages [62].
DNA methylation is a major epigenetic mechanism that has been shown in eukaryotes to play an important role for gene control, cell differentiation during development [60], embryonic development, chromatin structure, X chromosome inactivation, chromosome stability and genomic imprinting [58]. Methylation has also been suggested as an important molecular mechanism in the maintenance of memory [63].
The machinery involved in the methylation of DNA in mammals consists mainly of two components, a DNA methyl transferases(DNMTs) and a methyl-CpG binding proteins (MBD) [58]. DNMTs establish and maintain DNA methylation patterns, whereas MBDs read methylation marks. Both enzymes also interact with histone deacetylase to repress transcription [64].
The methylation of DNA is usually associated with the silencing of gene expression by directly blocking transcription regulatory factors from binding to their target sequences [57]. The proposed mechanism is that the methylation of DNA causes recruitment of binding proteins that recognize the methylated DNA and associate with histone deacetylase and chromatin remodeling complexes to cause the stabilization of condensed chromatin [65]. These binding proteins play a role in chromatin modification and remodeling and do not act in isolation; evidence has shown that they often interact with each other by forming large protein complexes [57].
Altered DNA methylation has been linked to many common human diseases [66]. Mutations in the DNMT genes have been shown to cause immunodeficiency, centromeric region instability, and facial anomalies syndrome (ICF) [67]. Defects in enzymes involved in epigenetic modification have been linked to various types of tumor formation and leukemia; elevated levels of DNMTs and MBDcontaining proteins have been observed in human tumors [66]. Both hyper- and hypo-methylation have been observed in cancer cells and the loss of methylation from repetitive regions of the genome results in genomic instability and is a hallmark of some tumors [66].
An increasingly popular methodfor DNA methylation analysis is to examine DNA extracts from peripheral blood to compare case and control samples [68]. Such studies of methylation status have been conducted in terms of various diseases. Johansson et al. [69] examined DNA methylation at 476,366 sites throughout the genome in peripheral blood samples from individuals ranging from 14 to 94 years of age and found that methylation plays an important role in the process of aging. Though there are numerous studies that use peripheral blood analysis to compare methylation profiles of controls vs. cases [70-72], a recent study insists on using caution when methylation analysis of peripheral blood is conducted for complex disorders [68]. The authors state that the results obtained from blood samples can be misleading as differences arise from the varying proportions of white blood cell types within the collected sample [68]. Still larger studies will have to be conducted to assess if changes in DNA methylation patterns are correlated with changes in disease endpoints, as inconsistent patterns may arise because of interpretation of results from different assays, and different sources of DNA. Multiple environmental factors like alcohol consumption, body mass index, smoking, folate intake, amongst others can impact methylation patterns within white blood cells depending on which loci are analyzed [73].
There is evidence that epigenetic mechanisms play a role in AD. Epigenetic modifications have been reported in disorders of synaptic plasticity and cognition [74]. The APP promoter is estimated to have a GC content of 72% and the rate of the CpG dinucleotides is five times higher than what is normally observed in other eukaryotic promoters [75]. Analysis of methylation status in healthy brain tissue failed to detect the presence of methylcytosines in the 460bp-275bp region of the APP promoter [75]. However, the 500bp upstream region showed brain tissue specific profiles of methylation that were associated with APP expression [75]. Studies have also suggested that age related demethylation may impact β-Amyloid deposition in the brain [75]. β-secretase (BACE1), and Prenesilin 1 (PSEN1) have also been shown to be regulated by methylation [75]. Genome wide analysis of CpG nucleotide methylation was performed at 27,000 loci within the frontal cortex, temporal cortex, pons and cerebellum of 387 individuals aged from 1 to 102 years. The authors report that some loci showed differential DNA methylation with increasing age. Methylation status has also been shown to vary amongst monozygotic twins since epigenetic differences arise throughout the lifetime of an individual [76]. Fragaet et al. [59] examined locus specific and global DNA methylation as well as histone acetylation patterns in a cohort of monozygotic twins. They found that during their early life, twins are epigenetically indistinguishable, but the overall epigenetic profile is different in older monozygotic twins.
In addition to methylation of cytosines, acetylation/deacetylation of histones has also shown to impact neurological disorders. The balance between Histone Acetyltransferases (HATs) and Histone Dacetyltransferases (HDACs) expression plays a role in neurological disorders, where the imbalance has been showing to play a roll in neuronal apoptosis [77]. Recently it has been found that malfunction of the HAT CREB-binding protein causes changes in chromatin acetylation status and this loss of function is associated with neurodegenerative disease [77]. The dysregulation of HATs can play a role in AD [78] and has been linked to clinical disorders, and inhibitors of histone acetyl transferases have been studied for use in treating neurodegenerative disorders such as Huntington’s disease, depression, and schizophrenia [74]. After initial cleavage of APP, γ-secretase activity generates β-Amyloid and an intracellular tail fragment. This intracellular fragment has been found to recruit the HATTip60, and may play a role in the expression of certain genes [74]. The protein HDAC6 positively correlates with tau burden, and a decrease in HDAC6 promotes tau clearance, making HDAC6 a key factor in regulation of tau protein levels [26]. HDAC inhibition could be an avenue thathas been suggested asa potential therapeutic approach for the treatment of a range of nervous system disorders [78].
Environmental factors such as oxidative stress and its impact on epigenetic modifications have been studied in human neuroblastoma cell lines. Gu et al. [79] found that oxidative stress increased intracellular β-Amyloid levels and BACE1 expression. Anincrease in BACE1 expression and a decrease in DNA methyltransferases was also correlated with demethylation of the BACE1 promoter region. Oxidative stress has also been observed to induce an increase in HAT expression and a decrease in HDAC expression. Caffeine has also been speculated as an environmental agent that is protective against AD progression, potentially as an epigenetic modulator [80]. In mammals, specifically monkeys, exposure to lead (Pb) at a young age and its impact on APP and BACE1 expression has been studied. The authors reported that exposure of lead caused an increase in APP, β-Amyloid , and BACE1 gene expression along with a decrease in DNA methtyltransferase activity and also observed higher levels of oxidative damage to DNA [81].
In contrast to the study of epigenetic regulation of genes related to AD, the field of epigenetic regulation of microRNAs (miRNAs) is still relatively new. MiRNAs are short noncoding RNAs that are involved in post-translational gene regulation by interacting with mRNAs and silencing genes. The mechanism of action involves inhibition of mRNA translation by binding the complementary target mRNA or by degrading the mRNA transcript. Recent studies have shown that epigenetic mechanisms, such as DNA methylation and histone modifications also regulate miRNA expression [82]. Further, a subset of miRNAs are known to manipulate the expression of important epigenetic regulators, including DNMTs, HATs and HDACs [82]. A feedback between miRNAs and epigenetic pathways works in tandem to regulate the entiregene expression profile [82]. MiRNAs are thought to control 30%of all protein coding genes in humans [83], by binding complementary target mRNA. It has been predicted that miRNAs affect genes that are involved in neuronal pathways that are altered in AD [84]. Studies have shown that miRNA profiles vary throughout the different regions of the AD brain [84], and miRNA-9, miRNA-125b, and miRNA-128 are upregulated in the hippocampus of the brain [85]. miRNA-9 is thought to target PSEN1, and an upregulation of PSEN1 could be associated with a decrease in miRNA-9 [85]. miRNAanalysis from human brain tissue of AD patients showed that as miRNA-107 levels decreased, BACE1 expressionincreased [86]. Peripheral blood studies showed up regulation of miRNA-181b, and miRNA-371 in AD patients in comparison to controls, suggesting that further studies could one day enable risk assessment of AD by analyzing miRNA expression profiles in peripheral blood [85]. Epigenetic regulation of miRNAs also needs to be considered in addition to regulation of genes involved in AD, as it was found that 13% and 28% of human miRNA genes are located within 3kb and 10kb of a CpG island respectively [87], suggesting that miRNA regulation can be affected via epigenetic modifications. Figure 1 gives an overview of how a network of multiple factors can play a role in AD etiology.
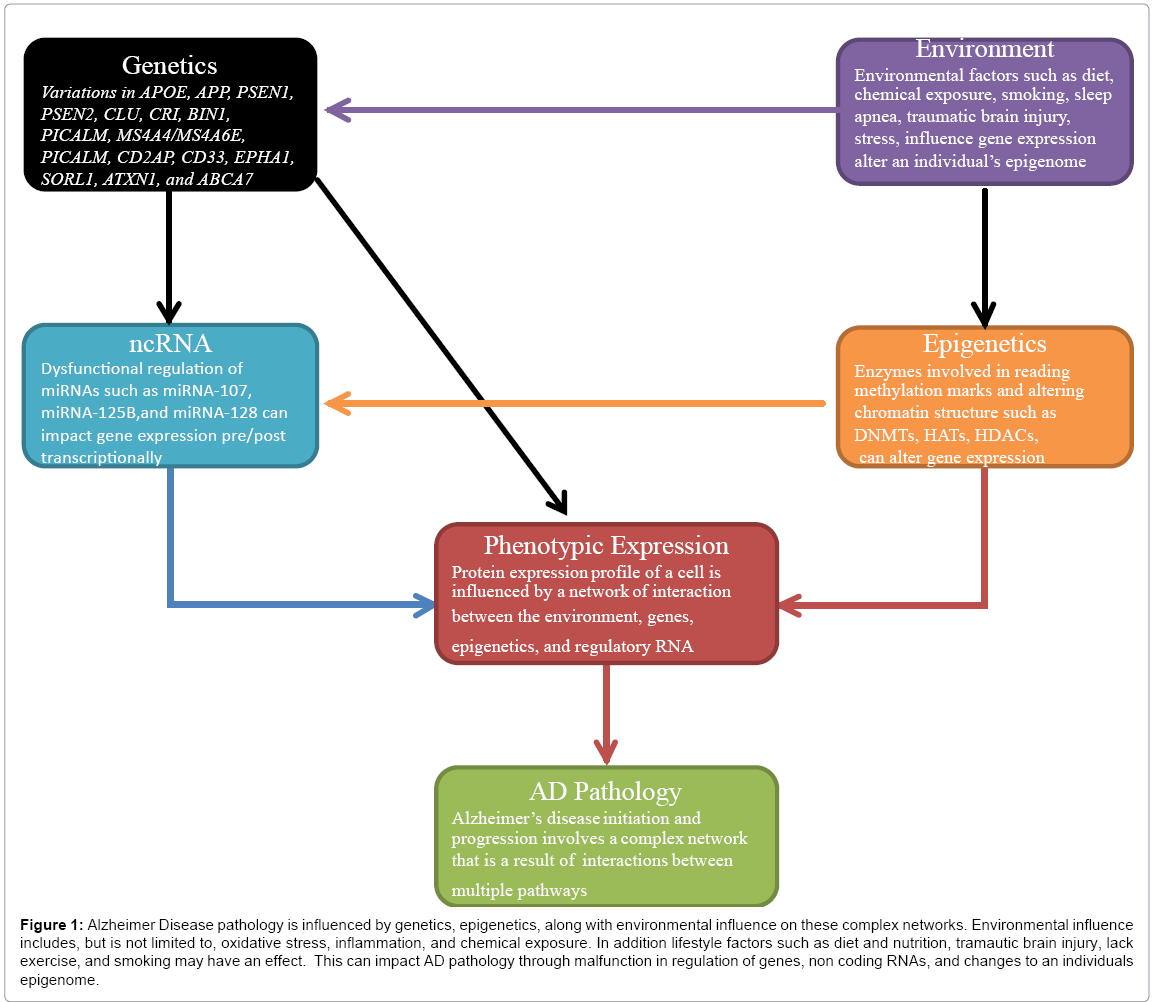
Figure 1: Alzheimer Disease pathology is influenced by genetics, epigenetics, along with environmental influence on these complex networks. Environmental influence includes, but is not limited to, oxidative stress, inflammation, and chemical exposure. In addition lifestyle factors such as diet and nutrition, tramautic brain injury, lack exercise, and smoking may have an effect. This can impact AD pathology through malfunction in regulation of genes, non coding RNAs, and changes to an individuals epigenome.
The genes involved in Familial Alzheimer’s Disease have been characterized and follow amendelianautosomal dominant inheritance pattern. In contrast, sporadic Alzheimer’s disease, or LOAD, is more complex and involves interplay between multiple loci and environmental factors. To date, Genome Wide Association studies (GWAS) have identified Single Nucleotide Polymorphisms (SNPs) within the human genome that play a role in AD. Even though GWAS have identified multiple genes that are associated with LOAD, a large proportion of the heritability currently remains unexplained. The impact of epigenetic mechanisms on AD risk and progression should be considered as we believe a portion of the remaining missing heritability can be explained via epigenetic effects within an individual and impacts the pathogenesis of AD. Oxidative stress (via traumatic brain injury, or sleep apnea, inflammation, etc....) can introducere active oxidative species, which causes integration of 8-oxoguanine within DNA, and lipid peroxidation that can impact the pathogenesis of AD. It is known that other environmental factors such as, nutrition, stress, exposure to chemicals can affect epigenetic modifications in the brain. These environmental factors may then impact gene expression and accelerate/decelerate mechanisms involved in neurodegeneration. This along with other lifestyle factors such as body mass index, smoking and folate intake can cause deviation from normal gene function by altering the epigenomic state of a cell. Regulation of gene expression can be altered if the epigenomic state of a cell is altered, as methylation pattern of DNA, acetylation/deacetylation of histones, the state of chromatin and non-coding RNAs, along with other proteins such as methyl-binding domains and transcription factors all work in concert to regulate gene expression. Population based studies also need to be conducted as methylation patterns for various loci across the genome have been shown to vary depending on the ethnicity of the individual [88]. Epigenetic regulation of microRNAs and other small non-coding RNAs such as PiwiRNA, have yet to be studied and may help to explain part of the missing heritability in sporadic Alzheimer’s disease. A lack of thorough knowledge remains on how epigenomics relates to the manifestations of neurodegenerative diseases. Overall understanding of this dynamic relationship between epigenomics, genetics, and the environment will certainly enhance our understanding of AD pathogenesis and possibly lead towards the development of novel therapeutic targets.
