Research Article - (2016) Volume 4, Issue 3
A series of new usnic acid ketamine compounds [1-8] and their oxime analogues [9-12] were synthesized by reacting (+)-usnic acid with various amines and subsequent treatment with hydroxylamine hydrochloride. They were evaluated on the human glioblastoma-astrocytoma cell line (U87MG) by a MTT assay for cell viability in vitro. The ketamine-derivatives (Schiff bases) show significant cytotoxicity on U87MG cells. A novel N-heterocyclic derivative (1,4-diazepine) showed an interesting tautomeric structure and displayed more activity on cancer cell line than (+)-usnic acid itself.
Keywords: Usnic acid derivative; DFT calculations; Glioblastoma cells; Cell viability
Usnic acid (2,6-diacetyl-7,9-dihydroxy-8,9b-dimethyl- 1,3(2H,9bH)-dibenzofurandione; C18H16O7) (Scheme 1) is a lichen metabolite and both the R(+) and S(-) isomers can be found in nature. (+)-Usnic acid and its derivative exhibit a wide range of biological activities [1] such as antibiotic [2], antiviral [3], apoptotic [4,5], analgesic [6], antipyretic [6] and anti-proliferative [7] activities. The toxicity in vitro and in vivo has been evaluated by Guo et al. [8] and hepatotoxicity but no general toxicity was observed. Antitumor activity of usnic acid was displayed for the first time by Kupchan and Kopperman [9] against Lewis lung carcinoma in mice. In addition, cytotoxicity activity of usnic acid and usnic acid-amine derivatives has been determined against a wide variety of murine and human cancer cell lines [4]. Although the cytotoxicity of usnic acid has been extensively reviewed, no data regarding the effects of usnic acid on glioblastoma cells (formally glioblastoma multiform, GBM) exists. GBM represents both the most common and most malignant variant among a number of primary brain tumors. In this study, usnic acid and ten derivatives of (+)-usnic acid were evaluated on a human glioblastoma - astrocytoma cell line (U87MG) by a MTT assay for cell viability. The N-heterocyclic derivative (1,4-diazepine) showed higher cytotoxicity on this cancer cell line than temozolomide (TMZ), which is the drug currently used against this cancer form.
The reaction between usnic acid and primary amines has been studied in a number of papers [10-21] leading to enamines and Schiff bases. One inherent problem with biological testing of usnic acid and some of its derivatives is a very low solubility in water [22]. The aim of this study is to synthesize new (+)-usnic acid derivatives with potential biological activities and better solubility. Ten derivatives of (+)-usnic acid were synthesized with ethylene diamine, series of aromatic amines and hydroxylamine.
(+)-usnic acid was obtained from Sigma (USA). Ethylene diamine, 2-aminobenzotrifluoride, 3-aminobenzotrifluoride, 4-aminobenzotrifluoride, 3-chloro-2-methylaniline, 5-chloro-2- methylaniline, hydroxylamine hydrochloride, benzylamine. Solvents and other reactants of high quality were purchased from Merck (Germany) and JT Baker (USA).
Melting points were measured on a Mettler Toledo (Switzerland) melting apparatus. IR spectra in KBr discs were recorded on a Nicolet 760 (USA) instrument. Preparative chromatographywas performed on a Shimadzu LC-20AP preparative system (Japan) with Agilent Prep SIL column (250 × 21.2 mm; 10 μm). High resolution mass (HRMS) measurements were recorded on a LCMS-IT-TOF Shimadzu (Japan) with an electrospray ion source (ESI). 1H NMR and 13C NMR spectra were measured at 500 and 125 MHz, respectively, on a Bruker AC-500 MHz (USA) or at 600 and 125 MHz on a Varian Inova instrument in DMSO-d6 or CDCl3 using TMS as reference. 2D NMR spectra, HSQC and HMBC were recorded according to standard protocols.
General methods
Procedure (A) for the preparation of compounds (1-2): A suspension of (+)-usnic acid (1 mmol) in a mixture of tetrahydrofuran and absolute ethyl alcohol (1:5; 12 ml) was treated with ethylene diamine (1.2 mmol, pure liquid), and heated to reflux for 4 hrs. with stirring. After the end of the reaction, the mixture was concentrated under reduced pressure. The obtained residue was purified by preparative chromatography.
(S,E)-6-acetyl-2-(1-((2-(((E)-1-((R)-6-acetyl-7,9-dihydroxy- 8,9b-dimethyl-1,3-dioxo-3,9b-dihydrodibenzo[b,d]furan-2(1H)- ylidene)ethyl)amino)ethyl)amino)ethylidene)-7,9-dihydroxy-8,9bdimethyldibenzo[ b,d]furan-1,3(2H,9bH)-dione(1): While solid (163 mg, 46%); mp. 186-187°C; Rf=0.51 (CH2Cl2/MeOH/NH4OH, 95:5:1); IR ν (cm-1) 3270, 3086, 2986, 1696, 1635, 1545; ESI-HRMS (m/z) calcd. for C38H36N2O12 (M+H)+ 713.2341, found 713.2343. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.65 (s, 3H, CH3-10); 1.94 (s, 3H, CH3-15); 2.51 (s, 3H, CH3-14); 2.62 (s, 3H, CH3-12); 3.95 (2H, CH2-16); 5.87 (s, 1H, H-4); 12.13 (s, 1H, OH-9); 13.06 (s, 1H, NH); 13.37 (s, 1H, OH-7). 13C NMR (125 MHz, DMSO-d6) δ (ppm) 7.4 (CH3-15); 18.1 (CH3-12); 30.9 (CH3-14); 31.6 (CH3-10); 42.4 (CH2-16); 56.3 (C-9b); 100.8 (C-6); 102.0 (C-2); 102.3 (C-4); 105.0 (C-9a); 106.3 (C-8); 155.7 (C-5a); 157.6 (C-9); 162.5 (C-7); 173.0 (C-4a); 175.7 (C-11); 188.7 (C-3); 197.4 (C-1); 200.8 (C-13).
(S)-9-acetyl-10,12-dihydroxy-5,11,12b-trimethyl-3,4-dihydro- 2H-benzo[2,3]benzofuro[4,5-e][1,4]diazepin-6(12bH)-one (2): Yellow solid (114 mg, 33%); mp 176-176.5°C; Rf=0.25 (CH2Cl2/MeOH/ NH4OH, 95:5:1); IR ν (cm-1) 3450, 2940, 1699, 1628, 1551; ESI-HRMS (m/z) calcd. for C20H20N2O5 (M+H)+ 369.1450, found 369.1450. For 1H and 13C NMR data see Table 2.
Procedure (B) for the preparation of ketamines 3-7 and (11): A suspension of (+)-usnic acid (1 mmol) in absolute ethyl alcohol (10 mL) was treated with amines (1.2 mmol, neat) and heated to reflux for 4 hr with stirring. After the end of reaction, the mixture was concentrated under reduced pressure. The obtained residue was purified by preparative chromatography.
(E)-6-acetyl-7,9-dihydroxy-8,9b-dimethyl-2-(1-(2- (trifluoromethyl)phenylamino)ethylidene)dibenzo[b,d]furan 1,3(2H,9bH)-dione (3): White crystals (204 mg, 42%); m.p. 150-151°C; Rf=0.36 (ethyl acetate/hexane/MeOH, 3:7:0.1); IR ν (cm-1) 3476, 2971, 1693, 1635, 1548; ESI-HRMS (m/z) calcd. for C25H20F3NO6 (M+H)+ 488.1321, found 488.1325. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.76 (s, 3H, CH3-10); 2.03 (s, 3H, CH3-15); 2.48 (s, 3H, CH3-14); 2.69 (s, 3H, CH3-12); 6.02 (s, 1H, H-4); 7.71 (m, 2H, H-4’, 6’); 7.86 (t, 1H, J=7.5, H-5’); 7.93 (d, 1H, J=7.5, H-3’); 11.92 (s, 1H, OH-9); 13.40 (s, 1H, OH-7); 14.84 (s, 1H, NH).13C NMR (125 MHz, CDCl3) δ (ppm) Ј (Hz) 7.5 (CH3-15); 20.8 (CH3-12); 31.3 (CH3-14); 31.9 (CH3-10); 57.7 (C-9b); 101.4 (C-6); 102.2 (C-2); 103.0 (C-4); 104.8 (C-9a); 108.3 (C-8); 122.9 (CF3, J=271.5); 127.0 (C-2’, J=30.8); 127.25 (C-3’, J=5.0); 128.8 (C-6’); 128.9 (C-4’); 133.1 (C-5’); 134.5 (C-1´); 155.8 (C-5a); 158.1 (C- 9); 163.6 (C-7); 173.5 (C-4a); 175.1 (C-11), 191.2 (C-3); 199.0 (C-1); 200.7 (C-13).
(E)-6-acetyl-7,9-dihydroxy-8,9b-dimethyl-2-(1-(3- (trifluoromethyl)phenylamino)ethylidene)dibenzo[b,d]furan-1,3 (2H,9bH)-dion (4): White crystals (326 mg, 67%); mp. 160-160.5°C; Rf=0.43 (ethyl acetate/hexane/MeOH, 3:7:0.1); IR ν (cm-1) 3471, 3086, 2925, 1698, 1628, 1549; ESI-HRMS (m/z) calcd. for C25H20F3NO6 (M+H)+ 488.1321, found 488.1323. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.71 (s, 3H, CH3-10); 1.98 (s, 3H, CH3-15); 2.53 (s, 3H,CH3-14); 2.65 (s, 3H, CH3-12); 6.01 (s, 1H, C4-H); 7.75 (s, 1H, H-2’); 7.80 (d, 2H, J=5.5, H-4’, 6’); 7.86 (t, 1H, J=5.5, H-5’); 11.92 (s, 1H, OH-9); 13.40 (s, 1H, OH-7); 14.84 (s, 1H, NH).13C NMR (125 MHz, CDCl3) δ (ppm) Ј (Hz) 7.5 (CH3-15); 20.6 (CH3-12); 31.3 (CH3-14); 31.9 (CH3-10); 57.7 (C-9b); 101.4 (C-6); 102.2 (C-2); 103.0 (C-4); 104.8 (C-9a); 108.4 (C-8); 122.9 (C-2’, J=3.8); 123.3 (CF3, J=272.3); 125.0 (C- 4’, J=3.3); 129.2 (C-6’); 130.4 (C-5’); 132.4 (C-3’, J=32.9); 136.8 (C-1’); 155.7 (C-5a); 158.1 (C-9); 163.7 (C-7); 173.9 (C-4a); 175.1 (C-11), 191.2 (C-3); 199.0 (C-1); 200.6 (C-13).
(E)-6-acetyl-7,9-dihydroxy-8,9b-dimethyl-2-(1-(4- (trifluoromethyl)phenylamino)ethylidene)dibenzo[b,d]furan- 1,3(2H,9bH)-dione (5): White crystals (317 mg, 65%); mp. 171- 171.5°C; Rf=0.48 (ethyl acetate/hexane/MeOH, 3:7:0.1); IR ν (cm-1) 3477, 2925, 1699, 1635, 1545; ESI-HRMS (m/z) calcd. for C25H20F3NO6 (M+H)+ 488.1321, found 488.1320. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.72 (s, 3H, CH3-10); 1.99 (s, 3H, CH3-15); 2.56 (s, 3H, CH3-14); 2.66 (s, 3H, CH3-12); 6.03 (s, 1H, H-4); 7.65 (d, 2H, J=7, H-2’, 6’); 7,89 (d, 2H, J=7, H-3’, 5’); 11.89 (s, 1H, OH-9); 13.40 (s, 1H, OH- 7); 14.94 (s, 1H, NH).13C NMR (125 MHz, CDCl3) δ (ppm) Ј (Hz) 7.7 (CH3-15); 20.9 (CH3-12); 31.5 (CH3-14); 32.1 (CH3-10); 57.9 (C-9b); 101.7 (C-6); 102.4 (C-2); 103.3 (C-4); 105.0 (C-9a); 108.6 (C-8); 123.8 (CF3-C4’, J=271.8); 126.4 (C-2’,6’); 127.2 (C-3’, 5’, J=3.8); 130.5 (C-4’, J=32.9); 139.5 (C-1’); 155.9 (C-5a); 158.3 (C-9); 163.9 (C-7); 173.9 (C- 4a); 175.4 (C-11), 191.4 (C-3); 199.2 (C-1); 200.8 (C-13).
(E)-6-acetyl-2-(1-(3-chloro-2-methylphenylamino)ethylidene)- 7,9-dihydroxy-8,9b-dimethyldibenzo[b,d]furan-1,3(2H,9bH)-dione (6): Bright white needles (380 mg, 78%); mp. 172-172.5°C; Rf=0.47 (ethyl acetate/hexane/MeOH, 3:7:0.1); IR ν (cm-1) 3483, 2923, 1699, 1635, 1542; ESI-HRMS (m/z) calcd. for C25H22ClNO6 (M+H)+ 468.1213, found 468.1218. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.73 (s, 3H, CH3-10); 1.99 (d, 3H, CH3-15); 2.27 (s, 3H, CH3-C3’); 2.45 (s, 3H, CH3-14); 2.66 (s, 3H, CH3-12); 6.01 (s, 1H, H-4); 7.38 (t, 2H, J=5, H-4’,6’); 7.55 (t, 1H, J=5, H-5’); 11.98 (s, 1H, OH-9); 13.40 (s, 1H, OH- 7); 14.69 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6) δ (ppm) 7.5 (CH3-15); 15.0 (CH3-C3’); 20.3 (CH3-12); 31.0 (CH3-14); 31.6 (CH3- 10); 56.7 (C-9b); 100.9 (C-6); 102.2 (C-2); 102.4 (C-4); 105.0 (C-9a); 106.5 (C-8); 125.8 (C-6’); 127.7 (C-5’); 129.1 (C-4’); 132.1 (C-2’); 134.5 (C-3’); 136.4 (C-1’); 155.6 (C-5a); 157.4 (C-9); 162.6 (C-7); 173.8 (C- 4a); 174.7 (C-11); 189.6 (C-3); 198.1 (C-1); 200.9 (C-13).
(E)-6-acetyl-2-(1-(5-chloro-2-methylphenylamino)ethylidene)- 7,9-dihydroxy-8,9b-dimethyldibenzo[b,d]furan-1,3(2H,9bH)-dione (7): Bright white needles (420 mg, 86%); mp. 234-234.5°C; Rf=0.46 (ethyl acetate/hexane/MeOH, 3:7:0.1); IR ν (cm-1) 3447, 2979, 1698, 1629, 1553; ESI-HRMS (m/z) calcd. for C25H22ClNO6 (M+H)+ 468.1213, found 468.1216. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.74 (s, 3H, CH3-10); 2.00 (d, 3H, CH3-15); 2.22 (s, 3H, CH3-C5’); 2.47 (s, 3H, CH3-14); 2.68 (s, 3H, CH3-12); 6.03 (s, 1l.H, H-4); 7.44 (d, 2H, J=8.5, H-3’, 4’); 7.55 (s, 1H, H-6’); 12.01 (s, 1H, OH-9); 13.41 (s, 1H, OH-7); 14.65 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6) δ (ppm) 7.5 (CH3- 15); 16.9 (CH3-C5’); 20.2 (CH3-12); 31.0 (CH3-14); 31.6 (CH3-10); 56.7 (C-9b); 101.0 (C-6); 102.2 (C-2); 102.4 (C-4); 105.0 (C-9a); 106.5 (C-8); 126.4 (C-6’); 128.3 (C-4’); 130.8 (C-3’); 132.5 (C-5’); 132.8 (C-2’); 136.2 (C-1’); 155.7 (C-5a); 157.5 (C-9); 162.5 (C-7); 173.8 (C-4a); 174.7 (C- 11); 189.6 (C-3); 198.1 (C-1); 201.0 (C-13).
Procedure (C) for the preparation of compounds (8-10): A solution of (+)-usnic acid or amine derivatives (0.25 mmol) and hydroxylamine hydrochloride (0.30 mmol) in 5% methanolic potassium hydroxide (6 mL) and distilled water (3 mL) was heated at 50°C under a nitrogen atmosphere for 2 hrs. The resulting solution was diluted with water (15 mL), acidified with 1N hydrochloric acid and extracted with ethyl acetate (3 × 15 mL). The combined extracts were concentrated under reduced pressure. The obtained residue was purified by preparative chromatography.
(E)-7,9-dihydroxy-6-((Z)-1-(hydroxyimino)ethyl)-8,9bdimethyl- 2-(1-(3(trifluoromethyl)phenylamino)ethylidene) dibenzo[b,d]furan-1,3(2H,9bH)-dione (8): White solid (67 mg, 67%); mp. 152-153°C; Rf=0.35 (ethyl acetate/hexane/MeOH, 3:7:0.1); IR ν (cm-1) 3379, 2926, 1692, 1630, 1546; ESI-HRMS (m/z) calcd. for C25H21F3N2O6 (M+H)+ 503.1424, found 503.1425. 1H NMR (500 MHz, CDCl3) δ (ppm) Ј (Hz) 1.68 (s, 3H, CH3-10); 2.00 (s, 3H, CH3-15); 2.35 (s, 3H, CH3-14); 2.53 (s, 3H, CH3-12); 5.89 (s, 1H, H-4); 7.75 (s, 1H, H-2’); 7.79 (d, 2H, J=5.5, H-4’, 6’); 7.85 (t, 1H, J=5.5, H-5’); 11.18 (s, 1H, OH-9); 11.50 (s, 1H, N-OH-13); 12.11 (s, 1H, OH-7); 14.91 (s, 1H, NH). 13C NMR (125 MHz, CDCl3) δ (ppm) Ј (Hz) 8.3 (CH3-15); 20.3 (CH3-12); 31.0 (CH3-14); 31.6 (CH3-10); 57.0 (C-9b); 101.3 (C-9a); 102.7 (C-6); 104.0 (C-2); 104.3 (C-4); 106.8 (C-8); 122.9 (C-2’, J=3.2); 123.6 (CF3-C3’, J=271.2); 124.6 (C-4’, J=3.5); 129.6 (C-6’); 130.7 (C-5’); 132.4 (C-3’, J=32.9); 136.8 (C-1’); 152.1 (C-13); 152.7 (C-7); 155.0 (C- 5a); 157.4 (C-9); 174.0 (C-4a); 174.4 (C-11), 189.8 (C-3); 198.7 (C-1).
(E)-2-(1-(5-chloro-2-methylphenylamino)ethylidene)- 7,9-dihydroxy-6-((Z)-1-(hydroxyimino)ethyl)-8,9b-dimethyl dibenzo[b,d]furan-1,3(2H,9bH)-dione (9): White needles (87.9 mg, 70%); mp. 172-173°C; Rf=0.37 (ethyl acetate/hexane/ MeOH, 3:7:0.1); IR ν (cm-1) 3414, 2921, 1698, 1630, 1538; ESI-HRMS (m/z) calcd. for C25H23ClN2O6 (M+H)+ 483.1323, found 483.1326. 1H NMR (500 MHz, DMSO-d6) δ (ppm) Ј (Hz) 1.78 (s, 3H, CH3-10); 2.00 (s, 3H, CH3-15); 2.20 (s, 3H, CH3-C5’); 2.45 (s, 3H, CH3-14); 2.66 (s, 3H, CH3-12); 6.00 (s, 1H, H-4); 7.43 (d, 2H, J=8, H-3’,4’); 7.53 (s, 1H, H-6’); 11.49 (s, 1H, N-OH-13); 12.09 (s, 1H, OH-9); 13.40 (s, 1H, OH-7); 14.65 (s, 1H, NH). 13C NMR (125 MHz, DMSO-d6) δ (ppm) 7.5 (CH3-15); 16.9 (CH3-C5’); 20.1 (CH3-12); 31.0 (CH3-14); 31.7 (CH3-10); 56.9 (C-9b); 101.3 (C-6); 102.2 (C-2); 102.5 (C-4); 104.4 (C-9a); 106.7 (C-8); 126.4 (C-6’); 128.2 (C-4’); 130.8 (C-3’); 132.4 (C-5’); 132.8 (C-2’); 136.3 (C-1’); 155.0 (C- 5a); 155.7 (C-13); 157.3 (C-9); 162.6 (C-7); 173.8 (C-4a); 174.5 (C-11); 189.8 (C-3); 198.5 (C-1).
(9bR)-2-acetyl-7,9-dihydroxy-6-((E)-1-(hydroxyimino)ethyl)- 8,9b-dimethyldibenzo[b,d]furan-1,3(2H,9bH)-dione (10): Yellow solid (94.6 mg, 72%); mp. 191-192°C; Rf=0.47 (ethyl acetate/hexane/ MeOH, 3:7:0.1); IR ν (cm-1) 3399, 3088, 2929, 1696, 1625, 1575; ESIHRMS (m/z) calcd. for C18H17NO7 (M+H)+ 360.1083, found 360.1084. 1H NMR (500 MHz, CDCl3) δ (ppm) Ј (Hz) 1.74 (s, 3H, CH3-10); 2.13 (s, 3H, CH3-15); 2.46 (s, 3H, CH3-14); 2.67 (s, 3H, CH3-12); 5.90 (s, 1H, H-4); 10.55 (s, 1H, N-OH-13); 11,73 (s, 1H, OH-9); 13.31 (s, 1H, OH-7); 18,77 (s, 1H, OH-3). 13C NMR (125 MHz, CDCl3) δ (ppm) 8.2 (CH3-15); 14.0 (CH3-14); 27.9 (CH3-12); 32.2 (CH3-10); 59.2 (C-9b); 97.4 (C-4); 98.9 (C-6); 104.0 (C-9a); 105.3(C-2); 109.3 (C-8); 153.1 (C- 5a); 157.8 (C-13); 158.0 (C-7); 180.2 (C-4a); 191.7 (C-3); 198.3 (C-1); 201.6 (C-11).
Preparative chromatography
High-speed fractionation was performed using a column with a high separation capacity so the obtained compounds could be achieved with high purity. The obtained residue was purified on a Shimadzu LC-20A using a silica column at ambient temperature. Separation was achieved using the mobile phases in Table 3 at a flow rate of 12 ml/min and was monitored using a PDA detector at 254 nm. In the purification step the yields were finally obtained in the range from 33% to 72%.
| Compound | Mobile phase |
|---|---|
| 1 | Chloroform - Methanol – Ammonia (95:5:0.1) |
| 2 | Chloroform - Methanol – Ammonia (95:5:0.1) |
| 3 | Ethyl acetate - hexane (5:5) |
| 4 | Ethyl acetate - hexane (4:6) |
| 5 | Ethyl acetate - hexane (3:7) |
| 8 | Ethyl acetate - hexane (4:6) |
| 9 | Ethyl acetate - hexane (4:6) |
| 10 | Ethyl acetate - hexane (4:6) |
Table 3: Mobile phases for preparative chromatography.
Biological studies
Cell culture and drug treatment: The biological studies were conducted in department of Pharmacology, School of Medicine, Kangwon National University, Chuncheon, Korea.
Human cancer cell line: U87MG glioblastoma cells were obtained from the Korean Cell Line Bank (KCLB), cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO BRL, Grand Island, NY, USA) plus 10% fetal bovine serum (FBS; Hyclone Laboratories, Inc.), 100 U/mL of penicillin, 100 mg/mL of streptomycin (GIBCO BRL, Grand Island, NY, USA). Cells were maintained at 37°C under a humidified 5% CO2 atmosphere.
Cell viability by the MTT assay: U87MG glioblastoma cells were plated into 12-well culture plates and were incubated in 10% serum culture at 37°C under a 5% CO2 / 95% humidified air incubator for 24 hrs. Cells were then incubated with TMZ, (+)-usnic acid or compounds 1-10 at indicated concentrations in serum free DMEM for an additional 24 hrs. The compounds were dissolved in DMSO and the level of DMSO in treatments did not exceed 0.1%. Cell viability was determined using the MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay, which yields a blue formazan product in living cells, but not in dead cells or their lytic debris. Then, cells were washed with PBS (phosphate buffered saline) and dissolved in DMSO (dimethyl sulfoxide, Sigma Aldrich). Absorbance was determined at 540 nm. The results were expressed as a percentage of MTT assay activity relative to the non-treated control. CC50’s of the specific compound was calculated, based on three concentrations using Origin software (www. originlab.com). The concentration of treating compounds at which 50% cell alive compared to non-treated samples was calculated from formula in corresponding to correlation curve formed individually, being considered as CC50’s of each compound. The experiments were repeated three times independently.
Theoretical calculations
The molecular geometries were optimised using the Gaussian09 suite of programs [23], Density Functional Theory (DFT) (Beckes [24] exchange and Lee, Yang, Parr [25] correlation term, B3LYP and basis set 6-31G (d,p) was used. The nuclear shieldings were calculated using the GIAO approach [25-27].
Synthesis
The reaction of (+)-usnic acid (2.0 equivalents) and ethylene diamine (1.0 equivalent) in a mixture of absolute ethyl alcohol and tetrahydrofuran (5:1) at reflux for 4 hr produced compound 1 only [28]. In our hands, the reaction of (+)-usnic acid (1.0 equivalent) and ethylene diamine (1.2 equivalents) gave compound 1 and the N-heterocyclic derivative 2 under the same conditions. The latter was predominant.
Compounds 3-5 were synthesized in absolute ethyl alcohol at reflux for 4 hrs. The reaction leading to compound 3 (42%) showed lower yield than compounds 4 (m-CF3; 67%) and 5 (p-CF3; 65%) (Scheme 1).
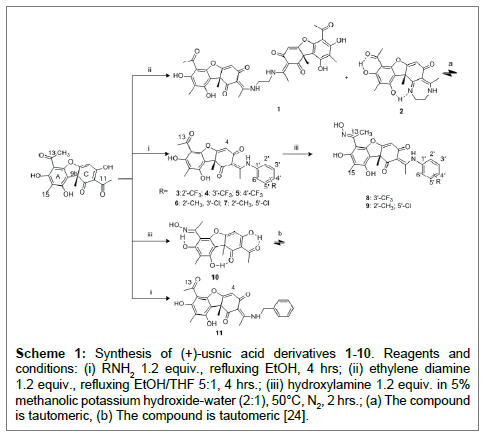
Scheme 1: Synthesis of (+)-usnic acid derivatives 1-10. Reagents and conditions: (i) RNH2 1.2 equiv., refluxing EtOH, 4 hrs; (ii) ethylene diamine 1.2 equiv., refluxing EtOH/THF 5:1, 4 hrs.; (iii) hydroxylamine 1.2 equiv. in 5% methanolic potassium hydroxide-water (2:1), 50°C, N2, 2 hrs.; (a) The compound is tautomeric, (b) The compound is tautomeric [24].
Under similar conditions 3-chloro-2-methylaniline and 5-chloro- 2-methylaniline gave compounds 6 and 7 in good yields, bright crystal needles 78%, 86%, respectively.
Treatment of compounds 4, 7 and (+)-usnic acid with hydroxylamine hydrochloride in 5.0% methanolic potassium hydroxide - water (2:1) at 50°C [10] resulted in good yields of the corresponding oximes (8, 9 and 10).
Structure elucidation and NMR chemical shift assignments
As shown by Kutney and Sanchez [11,14,15,19,20] does the reaction with primary amines occur at the carbonyl group C(11)=O leading to an enamine. This is confirmed in this study. Schiff base formation involves the amine attacking at C+=O. C-11 is part of a triketone system and therefore is more reactive than C-13 being part of an aromatic systems. This means that the Schiff base is formed at C-11.
The assignment of the NMR chemical shifts is central to the assignment of structures. Following the assignments of Ref. [19] it can be seen that the chemical shifts of the A-ring (Scheme 1) of mono substituted derivatives are very similar to those of usnic acid itself. The assignments of the other compounds are based on chemical shifts of usnic acid and this reasoning. For the benzene rings of compounds 3-7, C-F coupling constants are of great help in derivatives 3-5 and the resonances of the benzene rings of compounds 6 and 7 can then be assigned based on substituent effects.
The structure and assignment of compound 2 will be discussed separately as the structure is rather complex. Based upon the HSQC spectrum (Table 1) C-4 can be assigned unambiguously and the methylene carbons can be identified. Based on the HMBC spectrum (Table 1) long-range correlations from OH-7 confirm the assignment of C-6, C-7 and C-8. Knowing the assignment of C-6 the cross-peak from CH3-14 assigns H-14 as well as C-13. Knowing the position of C-8, cross-peaks from CH3-15 assigns H-15. The cross-peak from OH-9 confirms that the resonances in the HMBC spectrum overlap, although they are separate in the 1D spectrum at a lower field. H-4 helps to assign C-2 and C-4a. As H-10 and H-15 are now assigned, the remaining methyl signal must be those of H-12 and H-14. Crosspeaks from these assign C-13 and C-11. Cross-peaks from H-10 assign C-9a, C-4a and C-1. The latter is very important and the assignment is confirmed by cross-peaks to the CH2 protons. The chemical shifts in DMF-d7 are very similar to those in CDCl3 (Table 1) so we may assume that the structure is similar in the two solvents.
| Carbon | 1Ha | 13Ca | Long range correlations | 13Cb |
|---|---|---|---|---|
| 1 | 175.9 | 175.0 | ||
| 2 | 100.5 | 99.9 | ||
| 3 | 187.95 | 187.4 | ||
| 4 | 5.74 | 103.5c | 56.1; 100.5; 174.1 | 103.7 |
| 4a | 174.1 | 174.1 | ||
| 5a | 156.3 | 157.6 | ||
| 6 | 100.1 | 99.4 | ||
| 7 | 163.65 | 167.3 | ||
| 8 | 108.1 | 108.1 | ||
| 9 | 162.1 | 164.0 | ||
| 9a | 107.1 | 107.7 | ||
| 9b | 56.1 | 56.2 | ||
| 10 | 1.74 | 31.8c | 56.1; 107.1; 174.1; 175.9 | 33.0 |
| 11 | 167.7 | 172.6 | ||
| 12 | 2.67 | 27.5c | 167.7 | 26.8 |
| 13 | 200.4 | 199.8 | ||
| 14 | 2.60 | 31.2c | 100.1; 200.4 | 31.1 |
| 15 | 2.03 | 7.0c | 108.1; 163.65 | 8.4 |
| 16 | 3.8 and 4.2 | 50.0c | 48.9; 175.9 | 50.7 |
| 17 | 3.55 and 3.8 | 48.9c | 50.0 | 48.8 |
| OH-7 | 13.44 | 100.1;108.1;163.65 | ||
| OH-9 | 15.65 (br) | 100.1(vw)d;108.1(vw); | ||
| NH | 7.27 (br) |
Table 1: 1H and 13C chemical shifts of compound 2.
Deuterium isotope effects on chemical shifts have also been studied in order to support the structural assignment. The deuteriation is done by dissolving the compound in CH3OD and removing the methanol by rotary evaporation. This type of isotope effect is useful in the study of intramolecularly hydrogen bonded compounds [29,30]. The deuterium isotope effect at C-9 (0.6 ppm) is unusually large. Also C-11 shows large isotope effects although somewhat broad. The large effect at C-9 indicates a strong intramolecular hydrogen bond or an equilibrium system [31,32]. The former is in line with the large OH chemical shift of 15.6 ppm. Both the OH-7 and the NH resonance show splittings due to isotope effects.
Based on the above information a structure can be suggested as seen in Figure 2. However, upon cooling to -60°C two broad resonances appear in the 1H spectrum at 12.7 and 9.95 ppm integrating roughly 1:1. These are ascribed to NH resonances. Furthermore, the CH2 resonances merge to one broad resonance. In the 13C spectrum many resonances are now broad: C-1, C-4, C-9, C-10, C-11 and CH2CH2 indicating that an exchange situation is at hand, suggesting that two different tautomer’s exist as seen in Figure 1.
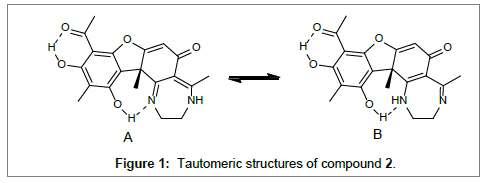
Figure 1: Tautomeric structures of compound .
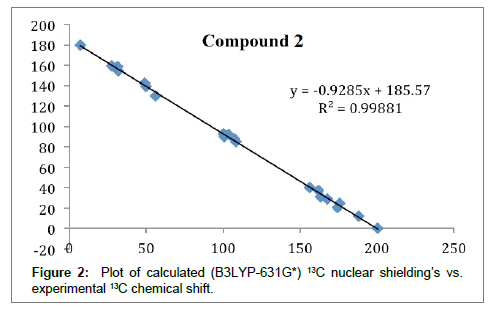
Figure 2: Plot of calculated (B3LYP-631G*) 13C nuclear shielding’s vs. experimental 13C chemical shift.
A comparison between calculated and experimental data for structure A shows a very good correlation (Figure 2). This correlation is not improved by adding data for structure B, so the former must dominate at ambient temperature. Kutney and Sanchez [10] found that using hydroxylamine hydrochloride in 5.0% methanolic potassium hydroxide - water (2:1) with the dibenzyl ether of usnic acid resulted in oxime formation at carbon 13 although a more logical result would have been at C-11 as amines react at this position (see previously). However, HMBC spectra confirmed that for 10 the reaction had occurred at C-13 (Scheme 1).
Biological activity
The MTT assay [29] was applied to evaluate the cytotoxicity of the derivatives on the human glioblastoma-astrocytoma cell line (U87MG) (Table 2).
| Compounds | Conc. (µM) | Cell viability(% control) 24 hrs | CC50(µM) |
|---|---|---|---|
| DMSO | 101.0 ±10.09a | ||
| (+)-Usnic acid | 10 50 100 | 114.0 ± 14.3 34.3 ± 8.1 20.0 ± 7.8 | 46.99 |
| 1 | >>100 | ||
| 2 | 10 50 100 | 102.3 ± 7.7 33.6 ± 7.0 11.3 ± 0.7 | 33.14 |
| 3 | >>200 | ||
| 4 | 10 100 400 | 102.4 ± 1.4 104.63 ± 10.7 75.71 ± 12.0 | >400 |
| 5 | >>200 | ||
| 6 | >>100 | ||
| 7 | >>200 | ||
| 8 | 10 100 400 | 83.2 ± 1.3 29.1 ± 6.6 8.0 ± 2.5 | 44.17 |
| 9 | >>100 | ||
| 10 | 10 50 100 | 102.7 ± 4.7 71.3 ± 6.7 48.3 ± 0.8 | 94.13 |
| TMZ | 100 400 | 83. 3 ± 14.5 61.0 ± 14.1 | >100 |
Table 2: Cytotoxicity of (+)-usnic acid and of ten derivatives 1-10 on the U87MG cell line.
The cytotoxicity of (+)-usnic acid and derivatives has been extensively reviewed [2,4,33,34], but there is no published data regarding its activity against U87MG cells, which was most commonly used to study glioblastoma multiform (GBM), the most aggressive brain cancer, according to WHO (World Health Organization). As seen in Table 2 the cytotoxicity of (+)-usnic acid and its ketamine-derivatives reduce the cell viability of U87MG significantly. Temozolomide (TMZ) is a DNA-alkylating agent approved by the U.S. Food and Drug Administration for treatment of the newly diagnosed GBM. However, the response of U87MG cell to TMZ is lower than that of (+)-usnic acid and its derivatives, whereas derivatives 2, 4 , 8, 10 and (+)-usnic acid show significantly cytotoxic effects on U87MG cells after 24 hr of treatment.
(+)-Usnic acid has a low solubility [22,34] in water (<250 μM, 25°C), ethanol (500 μM, 25°C) and organic solvents such as ethyl acetate or methanol. In this cytotoxicity study, TMZ, (+)-usnic acid and its derivatives were dissolved in DMSO, and derivatives 8 had a better solubility in DMSO and ethanol (100 μM, 25°C) than (+)-usnic acid. Thus, the dose of derivative 8 can be increased to 400 μM which significantly decreases the U87MG cell viability of 92% after 24 hr of treatment. In contrast, the chloro-2-methylaniline (6, 7, 9) derivatives have less activity on U87MG cells than the others. Compound 2 was the most potent usnic acid derivative against U87MG cells (CC50=33.14 μM and displayed dramatically higher activity than (+)-usnic acid or TMZ (Table 2).
The sensitivity of (+)-usnic acid is varying from one cell line to the next. The CC50 observed in U87MG cells after 24 hrs. (47 μM) is corresponding to previously published IC50 values in K-562 and DU145 cells (53 and 57 mM, respectively), but after 72 hrs. [4]. The time effects of (+)-usnic acid need to be addressed. In human keratinocytes (HaCaT) IC50 was estimated to 70 μM for (+)-usnic acid after 24 hrs. [20]. Both the (+) and (-) isomer of usnic acid are found naturally and the (-)-isomer shows generally a lower cytotoxicity compared to (+)-isomers [4,35]. Although a recent study relating to Mycobacterium tuberculosis showed similar activity for the two isomers [35]. A recent paper by Bruno et al. showed less cytotoxicity in rat myoblasts (L6 cells) of Schiff bases of usnic acid relative to the mother compound itself [19]. Our preliminary bioactivity data suggests that (+)-usnic acid and its derivatives could be potential cancer chemical therapy agent although a profound study is needed to examine the specificity of cytotoxicity on cancer cell lines as well as mechanism of activities in vitro and in vivo.
Condensation of (+)-usnic acid with amines and hydroxylamine gave a series of ketamine derivatives and its oxime derivatives. Of particular interest is the formation of a new type of compound 2. In addition, the cytotoxic activity of (+)-usnic acid and ketamine derivatives was evaluated using a MTT assay. The new compounds 2 and 8 were more active compounds in terms of cytotoxicity and they had an improved solubility. Compound 2 showed better activities (CC50=33.14 μM<46.99 μM) than (+)-usnic acid in this study. However, further investigations are needed to evaluate the biological activity of these compounds in relation to U87MG cancer cells and to improve the activity of usnic acid derivatives in vivo.
This work was supported by a research grant from the Institute of Drug Quality Control, Ho Chi Minh City, Vietnam.
