Research Article - (2014) Volume 0, Issue 0
Parts of the plant or the plant as a whole has been used in most parts of the world for the treatment of various ailments; either as topical applications to treat skin diseases or prepared into infusion, decoctions or even concoctions with other herbs and consumed to either alleviate pains or treat other diseases like malaria, pile, bacterial infective HIV etc. In an attempt to standardize this plant, the pharmacognostic studies were carried out on its stem bark. Preliminary processing of the plant material was done. The stem cuttings were debarked and the barks dried in an open air under shade. The macroscopical examinations were done. The dried plant materials was then powdered using morter and pistil. The anatomical sections and powdered samples of the plant parts were investigated for their microscopical profiles. These revealed the presence of phloem tissues, parenchyma cells, cork cells, calcium oxalate crystals, starch grains and secretory ducts in the powdered bark; while the anatomical sections revealed the presence of xylem tissues. A preliminary phytochemical screening revealed the presence of some phytochemicals. A co-chromatographic analysis revealed the presence of catechol (condensed tannins).
Keywords: Anatomical; Co-Chromatography Macroscopical; Microscopical; Monograph
The use of plants in ethno-medicine is continually being employed worldwide as a guide to the possible therapeutic potentials of a particular plant. For example, Fresh bark or leaves of the plant Detarium microcarpum are applied to wounds, to prevent and cure infections; cutaneous, subcutaneous parasitic infection. In Burkina Faso the fruit pulp is used for treating skin infections. A preparation of the fruits is taken against dizziness in Niger and Togo. In Senegal a mixture of the leaves of D. microcarpum, Sclerocarya birrea (A. Rich.) Hochst. and Acacia macrostachya Rchb. ex DC. Pounded in milk is considered very efficient for snakebites. In Benin a decoction of the leaves is taken to treat fainting and convulsions. In West Africa the roots are part of a medico-magical treatment for mental conditions, and for protection against bad spirits. In veterinary medicine the leaves and roots are used to treat diarrhoea in cattle in southern Mali, and in Benin to treat constipation. In Niger cattle are made to inhale the smoke of the leaves to treat fever. The plant as a whole is used in the treatment of arthritis, rheumatism, etc.; genital stimulants/depressants; leprosy; liver, etc [1]. In Mali the bark is also used to treat measles, nocturia, hypertension, itch and tiredness, while a decoction of the leaves or roots is taken against paralysis, meningitis, tiredness, cramps and difficult delivery. This helps the researcher to ultimately come up with ways in which such plant could be used in treating certain diseases and infections or develop drugs in the orthodox medical practice [2]. Despite the various medicinal uses to which this plant has been put into, it lacks scientific basis. Although previous work has been done on the fruits, fruit pulp, seeds, seed coats, leaves, either by way of screening its aqueous extracts against clinical isolates [3], investigating the response of some farm animals to diets containing the fruit pulp [4], comparing the biological actions of the chemical constituents of the leaves, stems and roots [5], there is no report or documentation made on the stem bark of Detarium microcarpum. The aim/purpose of this work is to come up with standards for the development of a monograph for the stem bark of Detarium microcarpum by evaluating the Pharmacognostic charactersters of its stem bark. This will serve as a reference point for the proper identification of this plant thereby contributing to the scientific world of research.
Collection, identification and preparation of the plant material
The plant material was collected from the wild in vegetation around the settlements in Giwa local Government Area located in Kaduna State, Nigeria. The collection was done at about 16:00 hrs of the day in December, 2008. The stem cuttings and twigs are the plant parts of interest thus, about 5 Kg of these were collected. In addition, two fresh young twigs of the plant material were also collected. These were stored in a glass jar under refrigeration and used within 24 hrs for the preparation of anatomical sections [6].
Another fresh twig of the plant material was collected and taken to the Herbarium of the Department of Biological Sciences A. B. U. Zaria for authentification/identification. A voucher number, 900676 was assigned to the plant sample on 17th December, 2008 by Mal. Musa and assisted by Mal. U.S. Gallah. Preliminary processing of the plant material was done. This includes elimination of undesirable materials and contaminants, washing to remove soil, sorting and cutting [2,6]. The stem cuttings were debarked and the barks dried in an open air under shade [2].The dried plant materials was then powdered using morter and pistil [2].
Macroscopical studies of the stem bark
The macroscopical examinations on the stem bark of Detarium microcarpum was carried out based on the features used in characterization of stem bark. It was described under the following headings:
Origin and preparation: whether whole or inner bark alone.
Size and shape: comprising the outer surface, inner surface, transverse surface as well as the nature of fracture
Organoleptic properties: Colour, odour and taste [2].
Microscopical studies of the stem bark
The microscopical studies were carried out as follows:
Microscopical Examinaton of Powdered Drug: About 5 g of the powdered stem bark were measured in a test tube and about 10 ml of chloral hydrate solution was added as a clearing agent, and the mixture was heated over water bath. The chloral hydrate solution also helps to dissolve starch grains, aleurone grains, plastids, volatile oils, and expand collapsed and delicate tissue without an undue swelling action upon the cell walls and distortion of the tissue [6]. Slides of these were prepared using dilute glycerine as the moutant. The various microscopic characters/features were observed under the compound light microscope.
Microscopical examinaton of anatomical sections: It involves fixing sections of the stem of the plant in paraffin wax. The procedure was carried out as follows:
Firstly, the material was fixed in Formalin Acetic Alcohol (FAA) for at least 24 hours. Thereafter, the following were carried out:
i. Rinsing of the plant material for 2 hours
ii. Dehydration by fixing the tissue in 30%, 50%, 70%, 95%, alcohol each for 2 hours duration. This is followed by further fixing in an absolute alcohol for two hours. This last step (i.e. in Absolute alcohol) is done twice.
iii. Cleaning or de-alcoholization: as soon as the tissue was removed from absolute alcohol, it was immediately fixed in Chloroform: Alcohol in the ratios 1:3, 1:1, 3:1, and finally in Absolute Chloroform. In each case, the tissue was allowed to stay for 2 hours and the one in Absolute chloroform was done twice.
iv. Impregnation of the Paraffin: little paraffin wax chips molten at 60°C were added; a little at a time by infiltration until chloroform no longer evaporates in which case impregnation process was expected to have taken place in molten paraffin wax at 60°C for 24 hours (overnight).
v. Solidification: The impregnated tissue was kept in a watch glass to properly embed the tissue in the paraffin wax. This was kept in this position until the time for sectioning.
vi. Sectioning: The prepared tissue was then mounted on a microtone set at 5 μ and then sectioned.
vii. Preparation of Slides: The sectioned tissue was placed on a clean slide and fixed on the slide using an adhesive (Glycerin and albumin)
viii. Staining Procedure: The sections were de-waxed in two stages using xylene for 5 mins each. They were then treated as follows:
• In absolute alcohol twice for 2 mins in each case, then 95%, 70%, 50% and 30% alcohol for 2 mins in each case and finally in distilled water for 2 mins.
• It was then stained in sarafin for 30 mins and rinsed in tap water for 1 min
• This was followed by de-staining using 70% Acid: Alcohol for 30 seconds.
• The de-stained tissue was then passed through 70%, 50%, and 30% Alcohol for 1 min in each case. Thereafter, it was washed in distilled water for 1 min.
• A counter staining was done using fast green for 1-2 mins, washed in tap water for 1 min and in distilled water for another 1 min.
• It was later passed through 30%, 50%, 70%, 95%, and in absolute alcohol twice for 1 min in each case.
• It was then passed through xylene in 2 stages for 5 mins in each case and finally mounted in D.P.X. which was a mixure of distrene (polystyrene), a pastilizer (tricresyl phosphate) and xylene or Canada balsam. It was then covered with cover slips.
At this stage, the permanent slide preparations were then ready for viewing for the microscopical characters [7,8].
Chemomicroscopic examination of the powdered stem bark: The presence of chemical substances eg tannins, starch and calcium oxalate crystals etc were tested for using the appropriate reagents as described below:
Test for Cellulose: Cellulose is the main constituents of plant cell wall [9]. They are responsible for the rigid cellular frame work [2]. The powdered stem bark was moistened with N/50 iodine and allowed to stay until nearly dry. Then 80% w/w sulphuric acid was added. A blue colour indicates the presence of cellulose in cell walls of epidermal cells and collenchymas cells of the cortex [2].
Test for Lignin: Lignin is a strengthening material which impregnates cell walls of tracheids, vessels, fibres, and sclerids of vascular plants Present in the greatest concentration in middle lamellae and in primary walls [2,9]. To a sample of the powdered stem bark, a few drops of phluroglucinol were added and allowed to stand for 2-3 mins until almost dry. A drop of concentrated hydrochloric acid was then added. A pink or cherry red color indicates the presence of lignified cells; particularly sclerenchyma cells [7].
Test for tannins: Tannins occur in mature dead cork cells [2]. Here, the three fractions were used. Firstly, 1 g of the crude extract was dissolved in a test tube using 70% methanol. Thereafter, a very dilute solution of ferric chloride was gradually added to the solution of the extract which resulted in blue-black colouration indicating the presence of gallitannins and ellagitannins (hydrolysable tannins). As more ferric chloride was added, the colour changes to olive-green showing that condensed tannins are also present in the powdered stem bark sample. The same procedure was repeated for the methanol and tannins fractions [2].
Test for mucilages: Mucilages are polysaccharide complexes of sugar and uronic acid; usually formed from the cell wall; insoluble in alcohol but swell or dissolve in water [9]. They are usually deposited in layers [2]. About 3 g of the powdered sample was moistened with a few drops of Ruthenium red. A reddish or dark pink coloration indicates the presence of mucilage in epidermal cells [2,7].
Test for starch: To about 5 g of the powdered sample, a few drops of N/50 iodine were added. A deep blue colouration indicates the presence of starch [2,9].
Test for fats and fatty oils: About 1-2 drops of Sudan IV were added to the powdered sample and gently heated over a water bath. The fatty substances in the powdered sample are stained orange-red or brick-red indicating the presence of fats and fatty oils [2].
Test for proteins: To about 3 g of the powdered plant sample was added a few drops of ninhydrin and the mixture was warmed gently over a water bath for about 5 mins. A pink, blue, violet or yellow colour (depending on the amino acid present) indicates the presence of proteins [2].
Test for calcium oxalate crystals: The powdered sample was first cleared using chloral hydrate solution. A little portion of the cleared sample was used to prepare a temporal slide with glycerine used as the moutant. When viewed under the light microscope, the crystal appears as bright structures of definite shape and size. 8% sulphuric acid was then added to the slide and viewed again. This time, the calcium oxalate crystals disappear. This confirms their presence in the original powdered sample [2].
Test for calcium carbonate: To about 5 g of the powdered stem bark sample was added 5 ml of acetic acid in a test tube. The calcium carbonate dissolves with effervescence indicating the presence of calcium carbonate. When 50% sulphuric acid was used, needle-shaped crystals of calcium sulphate gradually separate [2].
Evaluation of crude drug
Determination of Moisture Content by the “Loss on Drying Method”: The following procedure was carried out to determine the moisture content of the powdered stem bark of Detarium microcarpum
• An empty evaporating dish was heated to a constant weight, allowed to cool and placed in a desicccator.
• Thereafter 3 g of the powdered drug was weighed into the dish; the dish with its content were placed in an oven at 105°C and allowed to dry to a constant weight. This was achieved by checking the weight at 30 mins interval after an initial drying for about an hour. Two identical weights read off consecutively confirm a constant weight.
• The total loss in weight (i.e. the weight of the moisture content) was determined by subtracting the final weight of the dish and powder from the initial weight of the dish and the powdered stem bark.
• Finally, the percentage of the moisture content was calculated with reference to the initial weight of the powdered drug (stem bark) [2,10].
Determination of Total Ash Value: The objective of ashing vegetable drugs is to remove all traces of organic matter which may otherwise interfere in an analytical determination [9].
i. The following procedure was carried out to determine the total ash value of the powdered stem bark:
ii. An empty nickel crucible was heated in an oven at 105°C to a constant weight. This weight was accurately noted and the crucible was allowed to cool and kept in a desiccator.
iii. 2 g of the powdered plant bark sample was accurately weighed into the crucible.
iv. The crucible and its content were heated gently until it was moisture free and completely charred.
v. The heat was gradually increased until most of the carbon (black residue) was vaporized and then the heating was done strongly until the residue was free from carbon (i.e. almost white).
vi. The crucible and its content were then cooled, weighed and the weight accurately noted.
vii. The heating cooling and weighing was continued until the weight of the residue (ash) became constant.
viii. The weight of the ash was determined by subtracting the weight of the crucible from the final weight (i.e. weight of the crucible and the residue or ash).
ix. The ash value of the powdered drug (in percentage) was then calculated with reference to its initial weight [3].
Determination of acid-insoluble ash value: The following procedure was carried out to determine the acid-insoluble ash value of the powdered stem bark:
i. The crucible containing the ash from the Total-Ash-Value experiment was transferred to a beaker containing 25 ml of dilute hydrochloric acid.
ii. This mixture was boiled for 5 mins and filtered through an ash-less filter paper.
iii. The content of the beaker was rinsed with water through the filter paper. This washing/rinsing was repeated 3 times and each time, the washing was passed through the filter paper in a manner so that the residue is collected in the tip of the cone of the filter paper.
iv. The funnel was then dried along with the filter paper in an oven at 105°C.
v. A clean nickel crucible was heated and its weight was accurately measured and noted.
vi. The filter paper with the residue earlier dried in an oven was folded into a small cone and transferred into the crucible. The crucible and its content was heated gently until the filter paper was completely charred then the heat was applied strongly for a few minutes. The crucible with its content was cooled and weighed. This final weight was noted accurately.
vii. The weight of the residue (the acid insoluble ash) was determined by subtracting the weight of the crucible from the final weight. The acid insoluble ash value (in percentage) was calculated with reference to the initial weight of the powdered drug from the Total- Ash-Value experiment [4].
Determination of extractive values
Alcohol-soluble extractive value: This was done according to the following procedure:
Five grams of the powdered stem bark was accurately weighed into a 250 ml conical flask. 100 ml of 90% ethanol was added and the stopper replaced firmly. The flask and its content was shaken on a mechanical shaker for 6 hours and then allowed to stand for 18 hours. The mixture was filtered quickly. The weight of a clean flat-bottomed evaporating dish was accurately determined. 20 ml of the filtrate was poured into the tarred evaporating dish and evaporated to dryness on a hot plate. The weight of the residue was determined from the 20 ml extract by subtracting the weight of the evaporating dish from the final weight and the Alcohol-extractive value was calculated with reference to the initial weight of the powdered drug [10].
Water-soluble extractive value: The procedure of Alcoholextractive- value above was repeated using chloroform-water (0.25% v/v chloroform in distilled pure water), in place of ethanol as the extracting solvent. The Water-soluble extractive value was the determined [9].

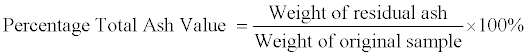
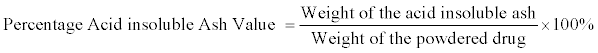
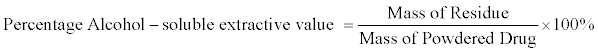
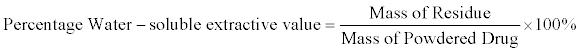
Phytochemical screening of the crude extract, tannin fraction and methanolic fraction
Standard screening tests of the crude extract and the other two fractions were carried out for the presence of various phytochemical constituents of the stem bark. This was done using standard procedures [1,11,12] (Table 1).
| Phytochemical | Test | Observation | Extract/Fractionsand Inferences | ||
|---|---|---|---|---|---|
| A | B | C | |||
| Carbohydrates | Molisch’s | A ring of purple colour | + | + | + |
| Fehling’s | Brick red precipitate of cuprous oxide | + | + | + | |
| Anthraquinones derivatives | Free anthraquinones (Borntrager’s test) | No change | - | - | - |
| Cardiac glycosides | Kellar-killiani | A reddish brown colour gradually turns blue | + | + | - |
| Legal | + | + | - | ||
| Kadde | A deep red colour, which fades to brownish yellow | + | + | - | |
| Saponins | Frothing | A honey comb froth which persists for about 10- 15 minutes | + | + | + |
| Haemolysis | presence of haemolysis | + | + | + | |
| Lieberman-Burchard’s–steroids | No Change | - | - | - | |
| Triterpenes | pink colour indicates the presence of triterpenoids | + | + | + | |
| Flavonoids | Shinoda | A pink or red colour | + | + | + |
| Sodium hydroxide | A yellow solution which becomes colourless in HCl | + | + | + | |
| Tannins | Lead sub-acetate | blue coloured precipitate | + | + | + |
| Gelatin test | Precipitates results | + | + | + | |
| Alkaloids | Mayer’s reagent | Turbidity or precipitation | + | + | - |
| Dragendorff’s reagent | Turbidity or precipitation | + | + | - | |
| Wagner’s reagent | Turbidity or precipitation | + | + | - | |
| Picric acid | Turbidity or precipitation | + | - | - | |
| Tannic acid | Turbidity or precipitation | + | - | - | |
Table 1: Phytochemical Constituents of D. microcarpum.
Chromatographic analysis
Co-chromatography using TLC plates: This was done following a column chromatography in which two unidentified fractions were obtained. The co-chromatography of the unidentified fractions obtained from the column chromatography was done alongside five (5) standard reference compounds. Co-chromatography is the chromatography of two or more samples together; especially in the identification of an unknown substance by chromatographic comparison with a known standard [11]. These reference compounds were as follows: catechol, gallic acid, phlorogucinol, pyragallol and quercetin (Table 2).
| Reference Compounds and Fractions | Colourswith Detection Reagents and Rf Values | |||||||
|---|---|---|---|---|---|---|---|---|
| Daylight | Rf | I2vapour | Rf | H2SO4 | Rf | FeCl3 | Rf | |
| Catechol | Light-yellow | 0.90 | Greyish-brown | 0.90 | Grey | 0.90 | Black | 0.90 |
| Gallic acid | Yellowish-brown | 0.78 | Greenish-brown | 0.78 | Grey | 0.78 | Purple | 0.78 |
| Phloroglucinol | Colourless | 0.83 | Carton-brown | 0.83 | Orange | 0.83 | Brown | 0.83 |
| Pyragallol | Light-brown | 0.85 | Brown-black | 0.85 | Purple | 0.85 | Black | 0.85 |
| Quercetin | Greenish-brown | 0.87 | Brown | 0.87 | Brown | 0.87 | Green | 0.87 |
| A2 | Colourless | Light-green | 0.83 | Brown | 0.83 | Green | 0.83 | |
| A3 | Colourless | Light-green | 0.83 | Brown | 0.83 | Colourless | ||
| A4 | Colourless | Colourless | Brown | 0.83 | Colourless | |||
| A5 | Colourless | Colourless | Brown | 0.83 | Colourless | |||
| A6 | Colourless | Colourless | Brown | 0.83 | Colourless | |||
| A7 | Colourless | Colourless | Brown | 0.83 | Colourless | |||
| A8 | Colourless | Colourless | Brown | 0.83 | Colourless | |||
| A9 | Colourless | Colourless | Brown | 0.83 | Colourless | |||
Table 2: The TLC Analysis of the Co- Chromatography.
Procedure: The solvent system was prepared and poured to a depth of about 3 cm in the chamber lined from inside with a filter paper to maintain the equilibrium of the mobile phase. The plates were activated in an oven at 120°C for 10 minutes. Standard solutions of the reference compounds were prepared by dissolving about 2 mg of each of the compounds in 2 ml of distilled water. The spots of individual reference compound solutions were applied (0.005 ml) as reference standards at a distance of 1-2 cm from each other with the help of a capillary tube. Five different spots of acetone fractions were also applied at the other end of the plates. The spots were then allowed to dry at room temperature. The glass plates were then gently placed inside the chamber and developed by ascending technique till the solvent front has moved about 10 cm. the plates were taken out and the solvent front were marked and the plate dried at room temperature. The characteristic colouration was firstly observed without any form of detecting reagent applied to the plates (daylight observation). The first plate was exposed to iodine vapour in an iodine glass chamber for about 10 minuites, the second plate was sprayed with 10% solution H2SO4 acid and heated at 100°C for about 5-10 mins while the third plate was sprayed with 5% solution of ferric chloride and also heated at 100°C for about 5-10 mins. The Rf values of each standard reference compound and that of the unknowns were calculated. These unknowns were then identified by comparing their colour and Rf values with that of the standard reference compounds [13,14]. The eluate cannot be monitored in the UV because of the strong UV absorbence of acetone [15].
Collection, identification and preparation of the plant material
A young twig of the plant was taken to the Herbarium of the Department of Biological sciences A. B. U. Zaria which was identified as Detarium microcarpum. A voucher number, 900676 was assigned to the plant sample on 17th December, 2008 by Mal. Musa and assisted by Mal. U.S. Gallah. A powdered material was obtained using morter and pestle.
Macroscopical studies of the stem bark
Based on the features used in characterization of bark, the stem bark showed the following as observed:
Origin was from the young and old stems of the plant.
Preparation: It was prepared whole i.e. consisting of all parts outside of the cambium (periderm).
Size was between 0.5-1.2 cm thick; the width were between 2-5 cm and length were between 14-18 cm long
Shape was generally curved and channeled shaped when dried. The outer surface has shades of milky-brown to grayish-brown with cracks or furrows running transversely while the inner surface has shades of brown to ox-blood red striation of colours running longitudinally with no furrows.
Fracture: It breaks with short granular fractures on the outside and short fibrous fractures with a few splinters on the inside.
Organoleptic properties
The powdered stem bark had the following characteristics:
Colour: light brown
Odour: Fairly aromatic
Taste: Astringent
Mircroscopical studies of the stem bark
Qualitative microscopical studies of the powdered stem bark revealed the following features:
Cork Cells: These are polygonal in shape. They occur at the epidermis in frequency of about 2-3 layers thick round the epidermis. They are about 11 μm in length and about 3.4 μm in width Figure 1.
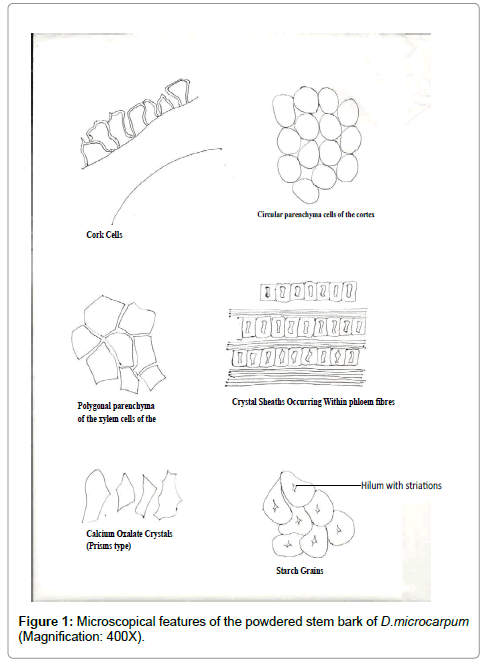
Figure 1: Microscopical features of the powdered stem bark of D.microcarpum (Magnification: 400X).
Circular parenchyma cells of the cortex: The diameter measured 21.6-23.8 μm in length. The cells occurred numerously in the cortex. Figure 1
Polygonal parenchyma cells of the phloem: The phloem parenchyma cells were polygonal in shape. The diameter measured 21.6-23.8 μm in length. They occurred numerously Figure 1.
Phloem Fibres: The phloem fibres were elongated in Shape. The Size ranges within 82-245 μm in length and 2.8-10.3 μm in width The Frequency of occurrence was moderate. They had tapering apices. The fibres were also slightly lignified Figure 1.
Calcium Oxalate Crystals: These crystals were the tetragonal type, measuring about 11-21 μm in length and 10-18 μm in width. The frequency of occurrence was moderate Figure 1.
Secretary Cells: Secretary cells contained mucilage, tannins or oils. Shapes were circular and quite few in occurrence Figure 1.
Starch Grains: The starch grains measured about 8-12 μm in diameter. Some were almost circular in shape while most were irregular. The position of Hilum was central. They occurred in aggregates was simple and the frequency of occurrence was occasional Figure 1.
Anatomical sectioning and examination
Transverse section of the stem: The transverse sections revealed the epidermal layer to be consisting of 1 to 2 rows of elongated parenchyma cells. Stomata were absent but lenticels were present at intervals. Trichomes were also observed on the epidermal layer. Next to these were 2 to 3 layers of collenchyma cells. The cortex comprises of parenchyma cells; (larger than those at the epidermis), secretary cells as well as sclerenchyma cells. This was followed by the phloem cells consisting of phloem fibres, phloem parenchyma and companion cells. The sieve tubes were not clearly differentiated. The cambium consists of thin walled elongated parenchyma cells in 2-3 rows. The xylem was observed to consist of xylem vessels, xylem fibres, xylem parenchyma and uniseriate medulary rays. The pith was the inner-most layer consisting of loosely packed almost circular parenchyma cells, with air spaces between them. Calcium oxalate crystals, starch grains, secretary cells and other cell inclusions were observed especially in the cortex Figure 2.
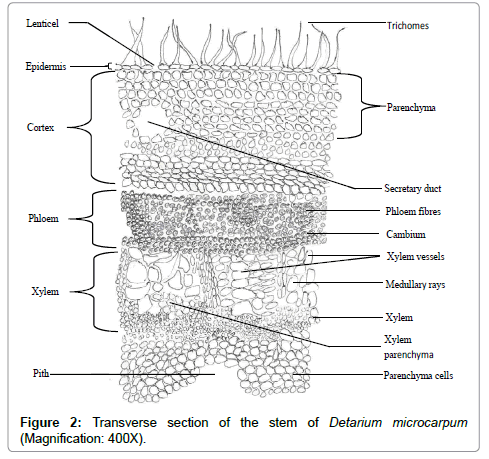
Figure 2: Transverse section of the stem of Detarium microcarpum (Magnification: 400X).
Longitudinal section of the stem: The longitudinal section revealed the various tissues seen in the transverse section. Furthermore, the type of xylem vessels became more detailed than in the transverse section. Vessels with spiral and annular thickening were well differentiated. Protoxylems, metaxylems and xylem fibres were noticeable. Calcium oxalate crystals were seen forming irregular rows on some phloem fibres Figure 3.
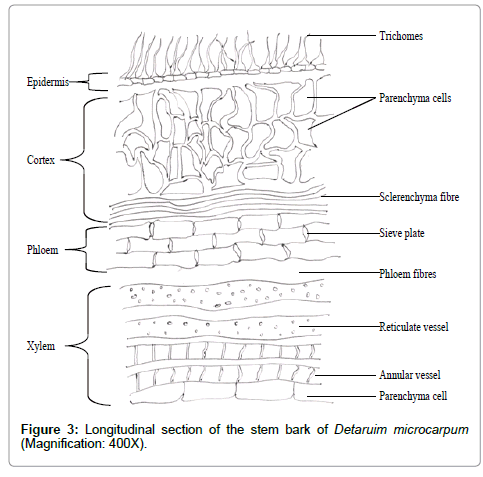
Figure 3: Longitudinal section of the stem bark of Detaruim microcarpum (Magnification: 400X).
Tangential section of the stem: The tangential section showed the tangential appearance of the various tissues observed with the xylem vessels pointing outwards. The phloem and xylem fibres were perpendicular to the pith. The medullary rays could be seen intermittently in between the xylem tissues Figure 4.
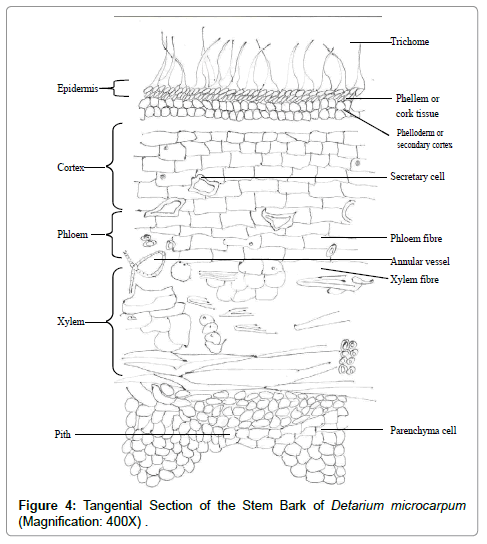
Figure 4: Tangential Section of the Stem Bark of Detarium microcarpum (Magnification: 400X) .
Co-chromatography
The co- chromatography revealed the presence of all the five reference standard compounds used and two distinct spots for the unknowns. The two unknowns were later confirmed to be catechol (condensed tannins). This is because, the Rf values and the characteristic colours they show when sprayed with the appropriate reagents corresponded to those of the reference standards used (Plates 1-3).
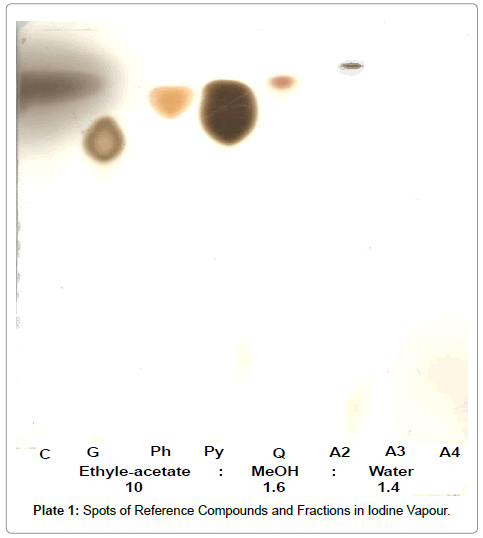
Plate 1: Spots of Reference Compounds and Fractions in Iodine Vapour.

Plate 2: Spots of Reference Compounds and Fractions when sprayed with 10% H2SO4.

Plate 3: Spots of Reference Compounds and Fractions of Extracts in Ferric Chloride.
The details of the macro and microscopical features and quantitative evaluative parameters were successfully established for the powdered stem bark of D. microcarpum. The features of the stem bark observed microscopically, e.g. the phloem fibres which were elongated tapering at the ends and slightly lignified, the calcium oxalate crystals which are tetragonal, several groups of secretory cells and ducts, rounded sclerids and circular/ irregular shaped starch grains are important diagnostic features of this plant (Detarium microcarpum). In addition to this, the anatomical sections (comprising the transverse, longitudinal and tangential sections) are important tools in the taxonomic classification of this plant as well as its proper identification.
The value of the moisture content, 5.1% w/w is an indication of the storage quality of the powdered sample. This is because; it is within the acceptable range [7] this is also indicative of its ability to retain its natural metabolites without any degradation. This is because when there is high moisture content in a particular drug sample in conjuctin with suitable temperature, enzymes will be activated and given suitable conditions, it ultimately leads to the proliferation of decay causing living organisms [2] therefore, low moisture content reduces enzymatic activity and does not support the growth of micro-organisms [2,10].
This also implies that the plant can be stored for a long period of time without any significant level of degradation.
The total ash-value (4.35% ± 0.38 w/w) is moderate which is also in indicating to some extent the amount of care taken in the preparation of the drug [2] This total ash value and the acid insoluble ash value (1.8% w/w) are parameters showing the amount of organic and inorganic materials in the powdered plant sample. The alcohol soluble and water soluble extractive values (22% w/w and 18% w/w respectively) indicate the extractive potency of the plant in alcohol and water. The higher value for the alcohol soluble extractive value, suggests that the use of the alcohol as the extraction solvent is a better choice for the polar metabolites present in the plant.
My sincere and profound gratitude my supervisors - Prof. H. Ibrahim for the immense academic guidance she offered, your words of encouragement has kept me focused and unflappable during the course of this research.
