Case Report - (2012) Volume 1, Issue 1
Paragangliomas are a type of neuroendocrine tumor derived from embryonic neural crest. They are mostly found in the adrenal glands (pheocromocytomas) and in the extra-adrenal paraganglia of the autonomic nervous system (extra-adrenal paraganglioma). Paragangliomas arising from the kidney or in the renal hilus are very rare and a preoperative diagnosis is hardly made. We present the case of a 44-years-old female with a 1-week history of feeling a mass in the right-upper side of the abdomen and no other symptoms. Computerized tomography (CT) demonstrated a round solid extra-adrenal mass located at the renal hilus. Laparotomy was performed with the intention of removing the mass and providing a histopathological diagnosis. Microscopic examination revealed a characteristic “Zellballen” organization and the presence of Sustentacular Cells S-100 positive. On these findings, extra-adrenal paraganglioma of the renal hilus was diagnosed histologically. The patient did not receive any further treatment other than surgery. The intervention was safe and effective. On a 20 months follow-up she results healthy and disease-free.
According to the World Health Organization classification of tumors, paragangliomas are a type of neuroendocrine tumor derived from embryonic neural crest. They are mostly found in the adrenal glands (pheocromocytomas) and in the extra-adrenal paraganglia of the autonomic nervous system (extra-adrenal paraganglioma).
Extra-adrenal paraganglioma usually arises in the abdomen, pelvis, thorax, neck and skull base [1].
Paragangliomas arising in the renal hilus are very rare and a preoperative diagnosis is hardly made. We present a rare case of nonfunctioning, extra-adrenal paraganglioma located at the renal hilus.
A 44-year-old female is admitted to the hospital with a 1-week history of feeling a mass in the right-upper side of the abdomen. There are no associated weight loss, abdominal pain, or history of pheochromocytoma commonly related symptoms, such as headache, palpitations, and diaphoresis, and no evidence of anemia, jaundice, and lymphoadenopathy. The patient had no known genetic predisposing factors for the developing of extra-adrenal paragangliomas or pheochromocytoma.
The patient was hemodinamically stable, with a pulse of 90 bpm, blood pressure of 140/80 mmHg (with no postural blood pressure drop). On examination, her abdomen was soft without peritonitis, with a firm and painless mass located in the right-upper abdominal quadrant; respiratory, cardiac and neurologic examinations did not show any pathologic findings. Abdominal ultrasound revealed a large mass in contact with the right kidney. Computed tomography (CT) demonstrated a round solid mass of 72x73x112 mm, located between the renal hilus above, the kidney parenchyma infero-laterally and the aortocaval axis medially. The mass had a high enhancement at its periphery. It looked compressing and dislocating the surrounding structures but it did not infiltrate them (Figure 1). Laparotomy was performed with the intention of removing the mass and providing a histopathological diagnosis. During surgical exploration the mass appeared soft and richly vascularized. It resulted located posterior to inferior vena cava, dislocating anteriorly the renal vein and posteriorly the renal artery. Because of the impossibility to obtain a complete exeresis of the mass sparing the renal vessels, a radical right nephrectomy plus right adrenalectomy and aortocaval lymphadenectomy was performed. The renal vein was dissected at its entrance in the inferior vena cava, and then repaired with a prolene suture 5/0. The ureter was dissected 5 cm far from its renal origin.
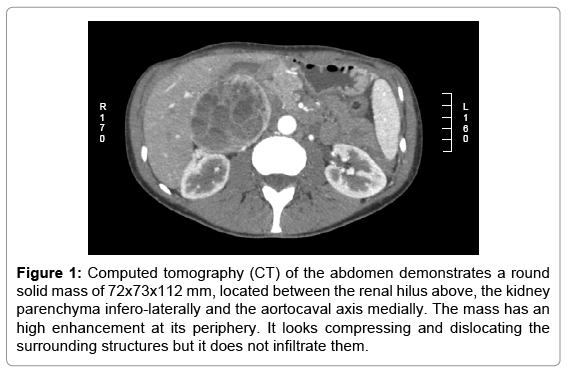
Figure 1: Computed tomography (CT) of the abdomen demonstrates a round solid mass of 72x73x112 mm, located between the renal hilus above, the kidney parenchyma infero-laterally and the aortocaval axis medially. The mass has an high enhancement at its periphery. It looks compressing and dislocating the surrounding structures but it does not infiltrate them.
A pathologist analyzed the specimen. The macroscopic analysis of the perirenal fat revealed a 9 cm diameter extra renal mass located close to the renal hilus but separated from the kidney and the adrenal gland. The mass looked soft, red-brownish, partially cystic and centrally hemorrhagic (Figure 2). No neoplastic emboli were noted inside the renal vein or in the minor vessels. No other anomalies were noted in the kidney, adrenal gland or perirenal fat tissue. Microscopic examination revealed neoplastic cells weakly positive for Neuron Specific Enolase (NSE), Synaptophysin and Chromogranin A. A proliferating cells fraction lower than 10% was found and occasional mitosis were observed. No necrosis was evidenced. Rare Sustentacular Cells S-100 positive were noted. The kidney, the adrenal gland and the perirenal fat tissue resulted negative for the presence of tumor cells or metastasis. On these findings, extra-adrenal paraganglioma of the renal hilus was diagnosed histologically.
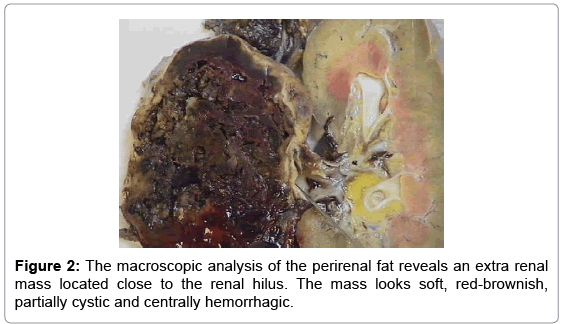
Figure 2: The macroscopic analysis of the perirenal fat reveals an extra renal mass located close to the renal hilus. The mass looks soft, red-brownish, partially cystic and centrally hemorrhagic.
The patient did not receive any further treatment. On a 20 months follow-up she results healthy and disease-free.
Paraganglioma located at the renal hilus is very rare. In literature we found only 14 cases reported up to 2007 (the research was managed on medline-pubmed entering as key-words: “paraganglioma”, “pheochromocytoma”, “renal hilum”). 6 reported cases of intra-renal paraganglioma and 8 cases of paraganglioma located in the renal hilus; only 4 of these 8 cases had non-functioning paraganglioma[2,3].
It is important to remember that, although WHO in 2004 had clearly defined the nomenclature and the difference between pheochromocytoma (that consistes only in intra-adrenal tumours) and paraganglioma, confused terminology persists and even in recent pubblications unprecise nomenclature makes sometimes difficult to distinguish pheochromocytoma and paraganglioma.
Paragangliomas usually occur sporadically, like the case we described, but they can also be associated with hereditary syndromes such as Von-Hippel Lindau disease (VHL) or, recently described hereditary pheochromocytoma-paraganglioma syndrome [4].
Pheocromocytomas and extra-adrenal paragangliomas can be associated with clinical evidence of epinephrine secretion (functioning tumors). Common symptoms are high blood pressure, pounding headaches, heart palpitations, flushing, nausea, and vomiting. Pheochromocytoma symptoms usually include paroxysms of extreme hypertension, accompanied by sweating, headache, and other autonomic disturbances. In the case of functioning tumors to make a diagnosis is surely easier but surgical treatment could be dangerous: in fact the surgical manipulation of the neoplastic mass may produce a risky hypertensive crisis.
There are no histological criterions to distinguish between functioning and non-functioning tumors [5]. In none of the nonfunctioning reported cases in literature preoperative diagnosis of paraganglioma was made [2,3]. On the other hand the absence of any clinical symptom related to catecholamines secretion leads the surgeon to take out safely the mass that is the only way to obtain a histopathological diagnosis.
The microscopic evaluation of the specimen revealed a classic “Zellballen” organization: well defined nests of cuboidal cells (“Zellballen”) separated by highly vascularized fibrous septa, the cells had an abundant granular cytoplasm and ultrastructurally they contained large number of cytoplasmatic neurosecretory granules [6]. The diagnosis was confirmed by the immunohistochemical evaluation, which revealed the absence of keratins but the presence of Sustentacular Cells S-100 positive. Keratins are absent in all extra-adrenal paragangliomas with exception of those arisen from filum terminale, on the other hand Sustentacular Cells S-100 positive are typically found in paragangliomas, and not in other endocrine neoplasms [7].
The clinical and histopathological distinction between benign and malignant paraganglioma is difficult and markers are lacking. Paragangliomas having higher level of S-100 positive cells seem to be associated with a benign clinical course [8]. On the other hand a malignant potential appears to be related to decreased immunohistochemical reactivity for a panel of neuropeptidases, an increased eneuploid content in DNA flow cytometry and to angiogenesis [9]. Actually only the evidence of metastasis seems to be the definitive criterion in differentiating benign from malign paragangliomas [7].
In the case of non-functioning extra-adrenal paraganglioma the treatment of choice and the best predictor of no recurrence seems to be the complete surgical removal of the mass [10].
