Research Article - (2015) Volume 3, Issue 1
Simple and selective extraction method of C- and N-terminal ions in ESI-MS/MS analyses, named isotopic differentiation protocol (IDP), was developed. It takes advantage of the mass-difference between the isotopologues after methyl and deuterated methyl ester formations at the C-terminal carboxylic acid or acetamide and deuterated acetamide formations at the N-terminal amines. This method distinguishes the N-terminal and C-terminal ions in principle to facilitate the de novo peptide sequencing. An application with non-ribosomal depsipeptide plipastatin revealed that a pair of the peptide and the corresponding methyl ester at the C-terminus was also applicable to IDP method giving spectrum with enough quality. Facilitating software named “MSMS Search Tool” was also developed.
Keywords: CID MS/MS peptide sequencing; Mass difference; Isotopic labeling; Facilitating software
IDP: Isotopic Differentiation Protocol; CID: Collision Induced Dissociation; PSD: Post Source Decay
MS/MS technology has contributed much not only in accelerating the peptide sequencing but also to scaling down of the experiments. MS/MS method utilizes the profiles of product ions after collision induced dissociation (CID) or post source decay (PSD) of the precursor peptide ions. Many algorithms, such as product ion mass finger print (PMF) [1-3] and peptide sequence tag (PST) methods, [4] as well as supporting software, such as Mascot [5], SPIDER [6], have been developed to facilitate the sequencings. It has been well established that the low energy CID of peptide ions predominantly affords the a- and b-ions (involving N-terminus) as well as y-ions (involving C-terminus) [7]. These ions exist simultaneously in an MS/MS spectrum, making the de novo peptide sequencings complex to require empirical skills for the researchers. Supporting software also refer the empirical data bases. However, those are poor to support the sequencings if the peptide involves unknown amino acid residues. These peptides are popular in non-ribosomal cyclic peptides [8]. To facilitate the de novo sequencing, James attempted to enhance the intensities of b-ion series selectively by introducing positively chargeable nicotinamide group at the N-terminal amino group [9]. Miyashita also emphasized the b-ions by derivatizations employing a series of benzoic acid derivatives at the N-terminus in a similar concept as James [10]. However, these methodologies might miss information from y-ions which complements those for a- and b-ion series.
James extended their methodology to isotopic labeling. The b-ion series were observed as doublets separated by 4 mass units while y-ions appeared as singlet, when the N-terminal amino groups were deuterium labeled with nicotinamides using a mixture of 1-([H4] nicotinoyloxy)- and 1-([D4]-nicotinoyloxy)succinimide esters [9]. They trapped both isotopologue ions with enough isolation width after derivatization. Lehmann et al. also have distinguished the y-ions in a similar concept by introducing 18O at the C-terminal carboxy group. This method requires the use of very expensive H2 18O as the solvent [11]. We herein report a simple discrimination between a, b-ions and y-ions by taking advantage of the mass difference between the isotopologues after the isotope labeling derivatization at either N- and C-terminus in inexpensive manners.
Methylation and deuterio-methylation of the C-terminal carboxylic acid
Figure 1 shows a fused LC-ESI-CID-MS spectrum (spectrum A) of O-methylated pentapeptide H2N-ALGLK-OCH3 (expressed as positive signals, target ion m/z = 515.4) and its [D3]-methyl isotopologue H2N- ALGLK-OCD3 (expressed as negative signals, target ion m/z =518.4). [H3]-Methyl and [D3]-methyl ester formation was performed by methanolic and deuterio-methanolic HCl, respectively, without remarkable peptide degradation. Since the latter case accompanied deuteration of amide protons because of using methanol-d4 as the solvent, those were replaced with protons again by adsorption on ODS SepPack® and the following elution with regular methanol. Non-labeled methyl ester could be subjected to LC-MS/ MS directly in contrast. The use of expensive methanol-d3 may allow us to skip these post treatments, but it is economically impractical. Low energy CID of peptides is known to provide the N-terminal fragments a-, b-ions and the C-terminal fragments y-ions predominantly [12]. Comparison of the MS/MS spectra between the isotopologues enabled us to classify the fragment ions into two groups; (i) common ions with similar intensities in both MS/MS spectra and (ii) ions with similar intensities but separated by three mass units. In principle, only the C-terminal y-ions show three mass unit differences (exactly 3.01883 mass units) between the isotopologues. Thus, ions (i) and (ii) are expected to be assigned as a-, b-ion series and y-ion series, respectively. Subtraction of the MS/MS spectrum of the [D3]-methyl ester from that of the [H3]-methyl ester would eliminate the common a-, b-ions to successfully extract the y-ions (spectrum B). Although the raw spectral data comprises very accurate masses in cases of TOF-MS, the product ion signals and their mass number had been converted to centroid bars and integer values, respectively, for simple operations. Dearing with more accurate mass values is expected to be more informative, thus that was considered when we developed the facilitating software as will be described in the next section. After the ions with negative intensities were slid -3 mass units, all the negative and positive signals were piled up to successfully emphasize the y-ions (spectrum C). The signals were colorized by origin of the isotopologue. Signals due to y-ions and its related signals are expressed with twotones with similar intensities. Since intensities of the product ions are not completely identical among the isotopologues, spectral subtraction sometimes would result to insufficient elimination of the common ions like that at m/z 242 in spectrum C. However, these signals are readily distinguishable, because these appeared as mono-tone signals. Noise signals also appeared as mono-tone one, such as that at m/z 296. N-terminal a-, b-ions could also be extracted selectively in the similar manner by changing the order of the process. The summary of those process was expressed like NMR DEPT-135 spectrum [13]; a,b-ion series with positive intensities and y-ion series with negative intensities. These procedures enabled us to visually assign the signals without empirical information. We named these data manipulations as “Isotopic Differentiation Protocol (IDP)”. Theoretically, critical limitations are expected for IDP especially when the mass numbers of bm-ion and yn-ion are identical and the mass numbers of bm’-ion and yn’- ion are separated by a particular mass units. (in the above case: 3 mass units). However, those occasions would rarely occur and we expected to overcome that by employing other mass difference between the isotopologues.
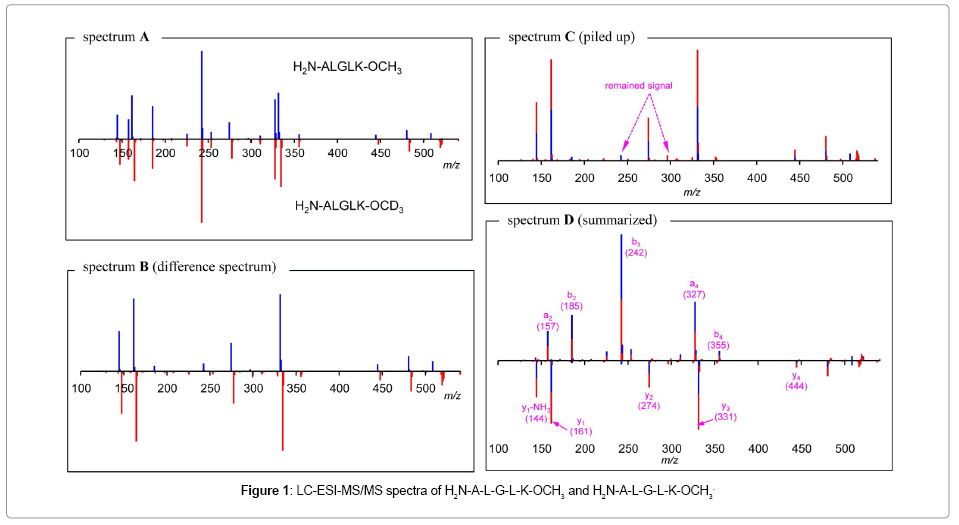
Figure 1: LC-ESI-MS/MS spectra of H2N-A-L-G-L-K-OCH3 and H2N-A-L-G-L-K-OCH3
Data processing application software “MSMS Search tool”
In the early stage, we manually processed the mass data tediously. After which, we developed an application software, the “MSMS search tool” to facilitate the analyses by performing the above data processing automatically after downloading a pair of the raw MS/MS spectral data of isotopically labeled and non-labeled peptides in ASCII format (Figure 2). Mass tables with peak intensities are also applicable (data delimited by tab). This program can deal with sub-integer mass numbers. Since the operation with integer numbers would result discontinuous at around 800-1000 mass units due to accumulation of decimal data in the real masses, analyses of peptides with more than 800 mass units will require more than one decimal place in the processing. Further accurate mass difference can be applied, that also requires exact calibration of mass numbers as well as enough data resolution. The program is supplied in public domain (Windows, http://www.t-kagaku.co.jp/seimeiken/msms/)[14].
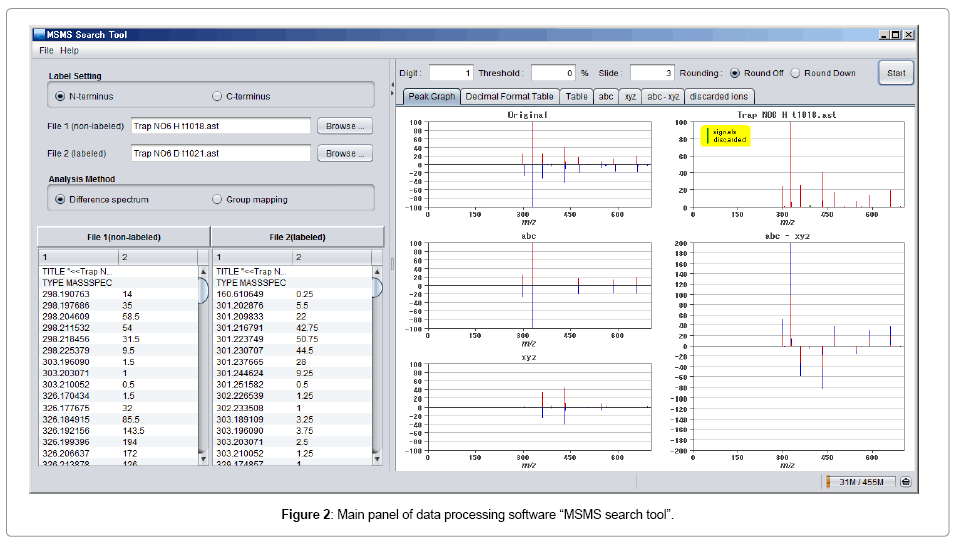
Figure 2: Main panel of data processing software “MSMS search tool”.
Scope and limitation of IDP
Applications with isotopic methyl esters at the C-terminus: The scope and limitations of the IDP with isotopic C-terminal methyl esters was next studied. Trials with several pentapeptides gave satisfactory results so far. Thus, we extended that to the sequencing of some synthetic decapeptides. Lys reside was designed at the C-terminus by taking the enzymatic digestion of proteins into account. When positively mono-charged ions (m/z 1019.5, 1022.5) of H2N-LAIYNAGVGKOCH 3(CD3) were chosen as the targets, the IDP successfully extracted the y-ion series (spectrum E in Figure 3). However, the a,b-ion series appeared complicated, due to non-assignable internal fragment ions. The CIDs employing doubly charged ions (m/z 510.3, 511.8) afforded clearer both y-ion and a,b-ion series after IDP, in spite the decreased number of a,b-ions (spectrum F). This protocol was found to be also applicable for mixture of isotopologues. The ESI-TOF of the isotopic mixture of H2N-LGIFNAGVGK-OCH3 and its deuteriomethylated isotopologue gave the mono-charged monoisotopic ions at m/z 989.59 and 992.61(spectrum G). Selective trapping of each ion followed by the IDP gave a quality spectrum (spectrum H).
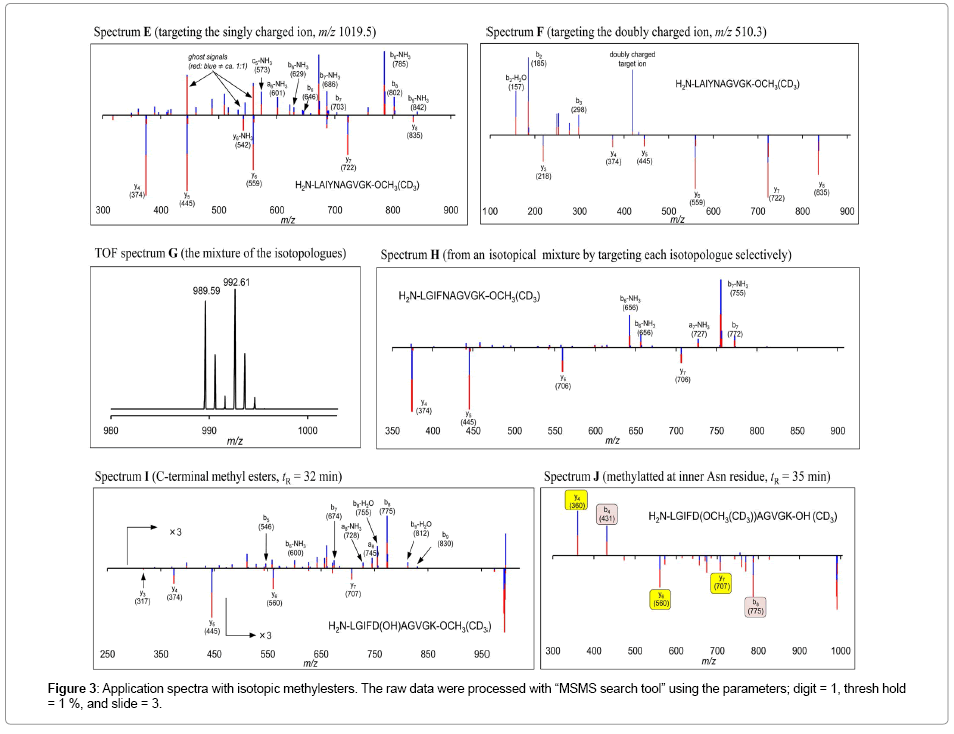
Figure 3: Application spectra with isotopic methylesters. The raw data were processed with “MSMS search tool” using the parameters; digit = 1, thresh hold = 1 %, and slide = 3.
This protocol became troublesome for peptides with acidic amino acid residues. For example, application of H2N-LGIFDAGVGKOH afforded H2N-LGIFD(OMe)AGVGK-OH which resulted to a complicated spectrum I. However, the reaction also gave the desired H2N-LGIFDAGVGK-OMe, providing sufficient IDP spectrum J.
As described, we demonstrated that IDP with isotopic methyl esters is effective in de novo peptide sequencing with MS/MS techniques. Our investigations also revealed limitations for this protocol; (1) deuteriomethyl ester formation accompanies deuteration of the amide proton which requires an operation to replace those with protons, (2) acidic amino acids, Glu and Asp, in the peptides hampered the analysis.
Applications with isotopic acetamides at the N-terminus: We next attempted the IDP after introducing the mass difference at N-terminus. James et al. has reported selective acylations of the terminal amine of the peptides by performing the reaction at pH 5.0 under aqueous conditions [9]. We investigated the IDP with acetamide groups. Although N-terminal selective acetylation using acetic anhydride has been reported by Noga et al. [15], 1-acetoxybenzotriazol and 1-deutrioacetoxybenzotriazole were chosen as the labeling reagents [16,17]. These are readily preparable, inexpensive, not volatile, stable for storage, easy to handle because it’s in powder form, and reactive enough for acetamide formation reactions. As expected, the James’s conditions successfully furnished acetamide group at the N-terminal amines (of H2N-LAIFNAGVGK-OH, H2N-LAIYNAGVGK-OH, H2NLGIFNAGVGK- OH, and H2N-LGIFDAGVGK-OH), without affecting the ε-amino groups in lysine residues at the C-termini. The present method has an advantage compare to the IDP with isotopic methyl esters because the reaction mixtures can be directly subjected to LCMS/ MS, while methyl ester formation required post treatments due to deuteration of amide protons as described (Figure 4).
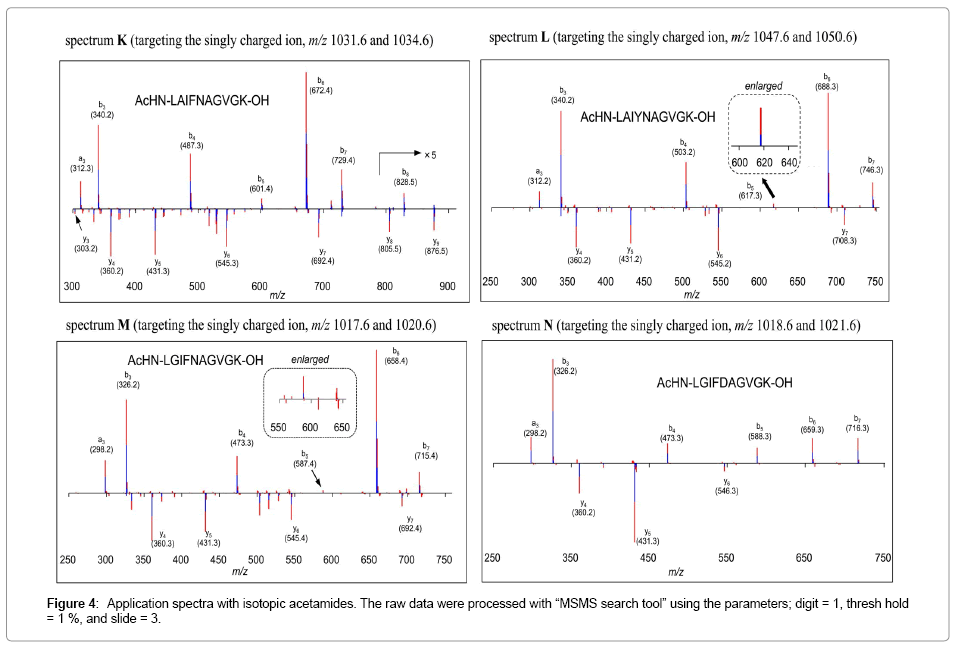
Figure 4: Application spectra with isotopic acetamides. The raw data were processed with “MSMS search tool” using the parameters; digit = 1, thresh hold = 1 %, and slide = 3.
Furthermore, the IDP with those samples made b-ion series more informative, while the IDP with methyl esters gave less number of b-ions. This method has led to slight qualitative deterioration in the y-ion series due to internal fragmentation for example in spectrum K. It seems to depend on the peptide sequence. So far, the degree of the deterioration seemed considerably lower than what we experienced in the previous IDP employing isotopic methyl esters. Tiny peaks m/z 617 and 587 in spectra L and M, respectively can be recognized as the sequence signals because those comprised of two-tone with similar intensities. This protocol seemed less sensitive in y-ion series in spite of positively chargeable amino group at C-termini.
An application for sequencing of depsipeptide, plipastatin: Finally IDP was extended to sequencing of amino acids in a depsipeptide plipastatin which we recently terminated the structural confusion with fengycin [18] (Figure 5). Treatment of plipastatin in methanol with aq NaOH afforded the corresponding seco acid and the corresponding methyl ester. Although those were not isotopologues, IDP of their MS/MS spectra gave the IDP spectrum O. Although some fragment ions were found as very weak signals, IDP extracted b- and y-ions were effectively with enough S/N level. Ions at m/z 843 and 948 could be assigned as invalid signals because of mono-tone signals. The positive signal at m/z 984 was the remained signal. Since these analyses differentiate the mass difference at the C-terminal groups, the y-ion series were quite reliable. The b8 and y3 ions were missing in the spectra, because of low energy CID which could not cleave the C-side of Pro residues [19]. Due to CID-trap-TOF, ions less than 400 could not be detected.
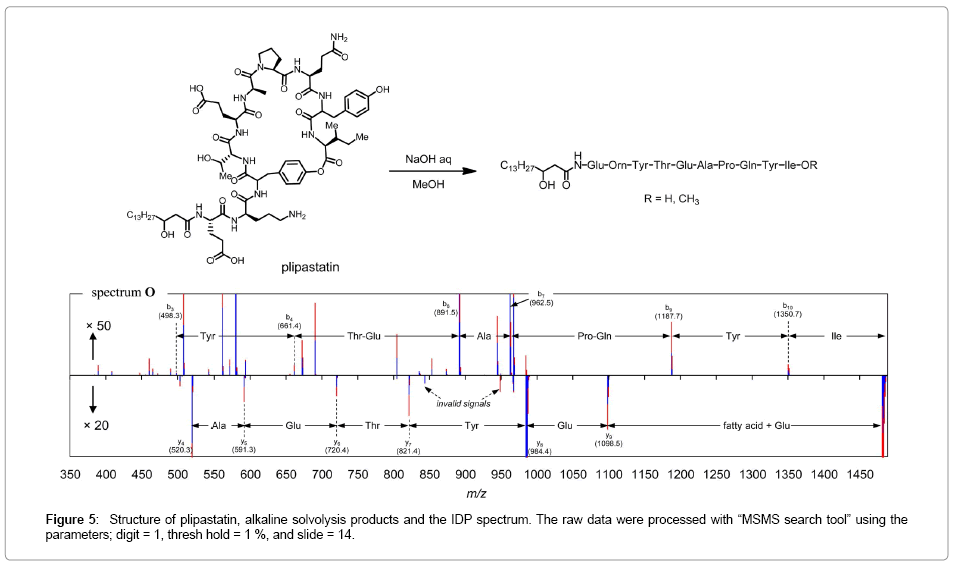
Figure 5: Structure of plipastatin, alkaline solvolysis products and the IDP spectrum. The raw data were processed with “MSMS search tool” using the parameters; digit = 1, thresh hold = 1 %, and slide = 14.
Application with isotopologues should be ideal for IDP; however, the present experiment successfully demonstrated its effectiveness for other type of differentiation.
As described, we developed an isotopic differentiation protocol (IDP) which extracts a,b-ions and y-ions selectively to facilitate de novo sequencing of peptides. Since IDP distinguishes these ions nonempirically, this method can be applied for the peptide involving unknown and unnatural amino acids. Mixture of isotopologues can be applied for IDP by trapping each isotopic ion selectively. The present study revealed that IDP with isotopic acetamides is more practical than that with isotopic methyl esters because of simple operations in the sample preparation as well as higher quality in the IDP spectra. Our application for the sequencing of plipastatin demonstrated that other mass difference such as between carboxylic acid and the corresponding esters are also applicable for IDP. Although we have not so far observed the considerable decrement in the sensitivity of the present protocol compare to the MS/MS with naked peptides, more sensitive and selective labeling groups are currently being explored in our laboratory.
Penta-peptides were synthesized in our laboratory and their purities were checked by LCMS. Deca-peptides were purchased from Hokkaido System Science Co. Ltd. Plipastatin was isolated in our laboratory from Bacillus subtilis H336B [18]. The LCMS and LCMS/MS were performed with a HITACHI NanoFrontier LD mass spectrometer equipped with a HITACHI 2100 high performance liquid chromatography (HPLC) pump, a HITACHI L-2400 UV detector, a HITACHI L-2300 column oven, and a HITACHI L-2200 auto sampler. The HPLC system was controlled by a PC using HITACHI EZChrom Elite. Inertsil ODS-3 (2.1× 33 mm, GL Science, Japan) was used for the HPLC column. The ESI-MS and ESI-MS/MS were performed with positive ion mode. The MS/MS conditions were not optimized, but the CID gain and CID time were set to provide analyzable numbers of fragment ion signals in each measurement.
Preparation of 1-acetoxybenzotoriazol
A mixture of acetic acid (208 mg, 3.5 mmol) and 1-hydroxybenzotriazol (HOBT, 475 mg, 3.5 mmol) in CH2Cl2 (5.0 mL) was stirred with WSCI·HCl ( 644 mg, 3.5 mmol) at -20°C for 1 hour. The mixture was poured into H2O (100 nL) and extracted with ether (50 mL×3). The organic layers were combined, washed with brine (50 mL) and dried with MgSO4. After filtration, the filtrate was concentrated under reduced pressure to give white solid. Recrystallization (AcOEt/ hexane) gave 1-acetoxybenzotoriazol (275 mg, 49%). The sample was observed as a kinetic mixture of N- and O-acetyl derivatives [16]. 1H NMR (500 MHz, CDCl3) δ 1.55 (3 H×0.4, s), 1.77 (3 H×0.6, s), 7.43 (1H+1H×0.4, m), 7.57 (1H, m), 7.79 (1H×0.6, dt, J = 1.0, 7.2 Hz), 8.01 (1H×0.6, dt, J = 7.2, 1.0 Hz), 8.07 (1H×0.4, dt, J = 7.2, 1.0 Hz), 8.41 (1H×0.6, dt, J = 7.2, 1.0 Hz).
Preparation of 1-deuteri-acetoxylbenzotriazole
In similar manner described in section 4-1, 1-deuteriacetylbenzotriazole (320 mg, 58%) was obtained employing acetic acid-d4 (192 mg, 3.0 mmol), HOBT (405 mg, 3.0 mmol), and WSCI·HCl (573 mg, 3.0 mmol).
Methyl ester formation
Thionyl chloride (5 μL) was added to a methanol solution (1.0 mL) of the peptides at 0°C. After 30 min, the mixture was subjected to LC-MS/MS directly. Since methanolyses of peptide bonds slowly proceeded, the reaction mixture could not be stored. The reaction mixture was concentrated in vacuo. Then the sample was adsorbed on ODS-SepPak® after dilution with H2O (1.0 mL) and washed with H2O. The following elution with methanol gave storable samples.
Deuterio-methyl ester formation
In similar manner described in section 4-3, the peptide was treated in methanol-d4. Since the reaction accompanied deuteration of the amide protons, the post treatment was required. After the reaction mixture was concentrated in vacuo, it was diluted with H2O (1.0 mL). Then, the sample was adsorbed on ODS-SepPak® and washed with H2O. The following elution with methanol gave storable samples.
IDP with a isotopic mixture of the esters
Methanol solutions of methyl- and dueterio-methyl esters were mixed by checking the quantity of the isotopologues to be almost 1:1 ratio. The LC-MS/MS were performed with CID window (3 msu) to trap the target ion selectively, while the window for the target ion was usually 5-10 msu.
Acetamide formation of the N-terminal of the peptides
The sample peptide (ca. 500 μg) was dissolved in acetate buffer (AcOH + NaOAc pH 5.0, 4.0 mL) and homogenized by pipetting. Acetoxybenzotriazole (ca 2.0 mg) was added to the solution and stirred at room temperature for 1.0 hour. In the case of deuterio-acethyl derivatization, deuterio-acetoxylbenzotriazole was added in place of acetoxylbenzotriazole. The reaction mixture was directly subjected to LS-MS/MS. The reaction mixture could be stored in the refrigerator without any post treatments.
Solvolysis of plipastatin
Plipastatin [18] (100 μg) in methanol (500 μL) with 1.0 M aqueous NaOH (30 μL) at room temperature for 30 min. The reaction mixture was subjected to LC-MS/MS directly without post treatments.
We would like to acknowledge Professor Katsuhiro Konno of Toyama University for his kind suggestions. Part of the present work was supported by JSPS Grant-in-Aid for Exploratory Research (24658104). The authors are grateful to Dr. Wilanfranco C. Tayone who kindly review and gave fruitful suggestions prior to the submission of the manuscript.
