Research Article - (2017) Volume 5, Issue 4
A number of significant herbal drug interactions have their origins in the alteration of cytochrome P450 (CYP) activity by various phytochemicals. In the present study, inhibition potential of Terminalia arjuna extracts and its constituent gallic acid (GA) and ellagic acid (EA) to cause herb-drug interactions through rat liver cytochrome enzymes, CYP3A4, and CYP2D6 was evaluated, a rapid RP-HPLC method was developed for quantitative estimation of GA and EA in the extract. In vitro safety of the extract and active components were evaluated through CYP450 inhibition method using pooled rat microsomes and high throughput fluorometric assay with CYP3A4 and CYP2D6. The binding mode and the molecular interaction of GA and EA within the CYP3A4 and CYP2D6 active site were demonstrated using an in-silico docking studies. From CYP450-CO Complex assay, the inhibitory potential of T. arjuna standardized extract (31.02 ± 2.24%), was found to be less than positive control. In high throughput ï¬uorometric assay, T. arjuna extracts exhibited higher IC50 values (48.06 ± 1.14-57.89 ± 2.15 μg/mL) compared to positive inhibitors and lower than GA (66.54 ± 1.04-83.84 ± 1.06 μg/mL), EA (69.47 ± 1.18-102.69 ± 2.87 μg/mL) on CYP3A4 and CYP2D6. Based on the inhibitory potential of test samples, it can be concluded that T. arjuna and its standardized bioactive molecules could produce weak interaction potential when co-administered with conventional medicines.
Keywords: Drug metabolizing enzymes; Terminalia arjuna; Safety evaluation; Gallic acid; Ellagic acid; Drug interaction potential
About 80% of people in developing countries still relay on traditional medicine their primary health care [1]. Elderly patients are using herbal drugs along with conventional prescription drugs due to their multiple illnesses [2]. When herbal drugs are co-administered with prescription drugs, there is an increasing risk of clinical treatment failures and adverse toxicity due to drug-herb interactions [3,4]. Terminalia arjuna (Roxb.) a deciduous tree of Combretaceae family, has been widely used in Indian system of medicine for cardiac ailments. Moreover, it is a vital component of common ayurvedic formulations available in India. It also possesses antioxidant, anti-inflammatory, antinociceptive and immunomodulatory activities [5-8] due to the presence of GA and EA along with other constituents.
CYP450 enzymes play a major role in the phase I oxidative metabolism of a wide range of pharmaceuticals [9]. Most of the pharmaceuticals are metabolized by or interact with only a small number of isoforms of CYP450. In this context, the most important forms are CYP2D6 and CYP3A4 [2,10,11]. Inhibition of CYP3A4 and/or CYP2D6 enzymes by herbal bioactive molecules may result in increased blood concentration of the target drug, which may lead to excessive accumulation and toxicity [12].
Various In vitro assays for measuring the activity and inhibition of CYP450 have been developed over the years [13]. HPLC-MS based assays are most commonly applied industrial standard method for CYP450 inhibition assessments. However, this method cannot be fit into high throughput format and may be difficult when huge number of samples needs to be tested. In this condition, fluorescence based CYP450 assays are much faster and more cost effective than HPLCMS assays [14,15]. In these assays, CYP450 oxidizes a pro-fluorescent molecule to a fluorescent product. This product can be directly measured using a fluorescent microplate reader. To our knowledge no data has been published on the interaction potential of T. arjuna plant extract or commercial product or its bioactive molecules on CYP450 mediated metabolism. Hence an attempt of systematic study has been taken to establish the safety issue through the interaction study with CYP450 including CYP3A4 and CYP2D6 isoforms.
Chemicals
All the chemicals and solvents for the research were of analytical grade. Vivid® CYP450 Screening Kit and Vivid® Substrates were purchased from Invitrogen Drug Discovery Solutions, USA. Vivid® CYP3A4 Red (Cat. no. P2856) and Vivid® CYP2D6 Blue (Cat. no. P2972). HPLC grade Methanol, acetonitrile and acetic acid procured from Merck (Mumbai, India) were used. GA and EA were purchased from Sigma Chemical Co, St Louis, MO, USA. 96 well black flat bottom polystyrene not treated microplate was obtained from Corning (Costar #3915, USA). Ketoconazole and quinidine was obtained as a gift sample from M/s Micro Labs Pvt. Ltd, Hosur, Tamil Nadu, India and M/s Trigenesis Life Sciences Pvt. Ltd, Bangalore, Karnataka, India respectively.
Plant material and preparation of test samples
T. arjuna, belonging to the family Combretaceae, holds a reputed position in both Ayurvedic and Yunani system of medicine. The bark of T. arjuna was collected from the central part of India (Madhya Pradesh) in the month of September 2013, and the authentication was done by the Prof. Jayaraman, Plant Anatomy Research Centre, west Tambaram, Chennai-600 045, Tamil Nadu. A Voucher specimen (IIISM/RES/ HER082) has been preserved in the Interdisciplinary Institute of Indian System of Medicine (IIISM) department, SRM University for future reference. Hydro alcoholic (70%) extract of coarsely powdered bark was prepared by cold maceration method, the extract was filtered and concentrated under reduced pressure by Rotovac (Buchi Rotavapor® R-210). The extract and the bio-marker compounds GA and EA were dissolved in ethanol and dimethyl sulfoxide (DMSO) for the experiments.
Standardization of the plant extract by RP-HPLC
Quantitative analysis of GA and EA were performed using Reversephase high performance liquid chromatography system (RP-HPLC). The HPLC system consisted of a water 600 (Milford, MA, USA) binary HPLC pump, a Prominence -7725i injection valve (USA) with a sample loop of 20 μL, a water 2489 UV-visible dual wavelength detector and the max-plot containing the peaks were obtained using Lab solutions software. A phenomenex-Luna (Torrance, CA, USA) C18 column (250 mm × 4.6 mm, 5 μm particle size) was used as stationary phase. The isocratic mobile phase consists of methanol: acetonitrile: 1% of acetic acid in water (40:15:45) was used with a flow rate of 1 mL/min and the samples were injected using Hamilton Microliter (Switzerland) syringe into a 20 μL injection loop, the reference standards GA and EA was detected at 280 nm and 254 nm respectively. 1 mg/ml of the reference standards were dissolved in methanol and this stock solution was subsequently diluted to prepare solutions with concentrations in the range of 10-100 μL/mL of GA and EA separately. Exactly weighed test samples (1 mg/mL) were sonicated with methanol for 30 min and filtered through Whatman NYL 0.45 μ syringe filter. An aliquot of 20 μL of test solutions was injected into the HPLC system at an interval of 10 min. the analysis was repeated for three times by comparing and interpolating the extract peak area with that of standards from the calibration curve.
Preparation of rat liver microsomes
Based on the method labeled by Tang et al. [16], Male wistar rats (200-250 g) were used for CYP450-CO Complex assay. The present study was approved by the Institutional Animal Ethical Committee (IAEC151/2015), SRM College of Pharmacy, SRM University. Briefly the rats were sacrificed by cervical dislocation method and abdomen was opened, 1.15% cold KCl solution was injected into the liver portal vein until the liver turned into khaki color. Filter paper was used to absorb water and the liver tissue was rinsed with the volume of cold 50 mM Tris-HCl buffer (50 mM Tris, 50 mM KCl, 0.5 mM EDTA, pH 7.5) in three times to make homogenates. The homogenates were centrifuged (Micro Centrifuge-5430R, Eppendorf AG, Hamburg, Germany) at 10000 rpm for 30 mins at 4°C to remove cell debris, nuclei, mitochondria. The supernatant was transferred to fresh centrifuge tubes and centrifuged at 10500 rpm for 60 min at 4°C. The resulting pellet was resuspended in storage 0.2M sucrose into cryovials. All processes were performed 0-4°C. Fractions were immediately frozen at -80°C and used within a week. Protein concentrations were determined by modified biuret method (Multiskan™ GO Microplate Spectrophotometer, Thermo Scientific, Waltham, MA, USA) using bovine serum albumin as standard.
CYP450-Carbon monoxide (CO) complex assay
Inhibitory activity of T. arjuna was performed with rat liver microsomes in 96 well microplate, based on method described by Ponnusankar et al. [17]. The concentration of P450 was calculated using the formula;
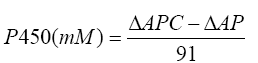
Where, ΔAPC is the absorbance difference of the PC sample, and ΔAP is the absorbance difference of the P sample. The percentage inhibition was calculated using the following formula.
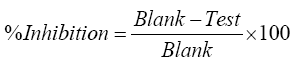
High throughput fluorometric assay of CYP3A4 and CYP2D6
High throughput screening (HTS) assays were performed in black 96- well microplates. Fluorescence readings were obtained on BioTekFLx 800 fluorescence microplate reader (Bio Tek, US) using appropriate excitation/emission (λ) wavelength. The assay was performed according to a protocol provided by Invitrogen Drug Discovery Solutions, USA. Respective positive controls were used for different isozymes; concentration of enzymes and substrate has been described in Table 1. All measurements were performed in triplicate. Product formation from the fluorogenic probes were determined from the fluorescence data at seven different concentrations of the inhibitors and test. IC50 values were determined by using the following formula.
| Experimental conditions/ CYP isoforms | CYP3A4 | CYP2D6 |
|---|---|---|
| Substrate concentration | 3 µM | 10 µM |
| Concentration of fluorescent standard | 100 µM | 100 µM |
| Master Pre-mix screening concentration | 5 nM | 10 nM |
| Positive inhibitor | Ketoconazole | Quinidine |
| Concentration of inhibitor | 10 µM | 1 µM |
| Incubation time | 30 min | 30 min |
| Excitation wavelength | 530 nm | 360 nm |
| Emission wavelength | 590 nm | 460 nm |
Master Pre-mix (recombinant human Cytochrome P450, glucose-6-phosphate and glucose-6-phosphate dehydrogenase)
Table 1: High throughput fluorescence inhibition assay experimental conditions.
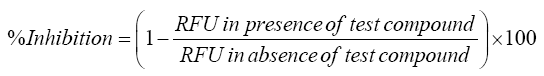
Where, RFU is Relative fluorescence unit.
Molecular docking analysis
Mode of inhibition of GA and EA on these CYPs was further assessed by molecular docking analysis with the software Glide (Schrodinger Inc. USA). Glide is a ligand docking program for predicting proteinligand binding modes and ranking ligands via high-throughput virtual screening [18]. Structures were downloaded from PubChem using chemical names. The compounds energy was minimized with various conformers, tautomers and ionization states using OPLS 2005 force field in LigPrep module in Schrodinger. Crystal structure of CYP3A4 and CYP2D6 were obtained from protein databank (PDB ID: 2F9Q and 1TQN) respectively. Using protein preparation wizard in Schrodinger module, protein was prepared for docking. In this step protein was minimized, assigning bond order, creating disulfide bond and water residues were removed. Those interacting water molecules with residues with protein and heteroatom were kept for docking studies. Then protein was refined by assigning H-bonds and minimization at OPLS 2005 force field. The glide grid was generated around the cocrystallized ligand heme compound in crystal structure of CYP3A4 and CYP2D6 using receptor grid generation module. Using extra precision (XP) docking algorithm of Glide module, GA and EA docked with CYP3A4 and CYP2D6.
Statistical analysis
All data are presented as mean values standard error mean (S.E.M) of three replicates if not stated otherwise. The results were subjected to one way analysis of variance (ANOVA) and Dunnett's multiple comparison test was performed by fixing the significance level at P<0.05 and above. IC50 values (concentration required to cause a 50% inhibition in enzyme activity) were obtained using mean enzyme activity versus inhibitor concentration curves using GraphPad prism Version 5.01 (GraphPad Prism Software Inc., USA).
Standardization of T. arjuna extract and its bioactive molecules by RP-HPLC
The concentration of bioactive markers, GA and EA was determined through RP‐HPLC using the isocratic condition and eluted through a run time of 10 min. The separation was performed with the mobile phase consisting of methanol, acetonitrile, 1% of acetic acid in water in the ratio of 40:15:45 with a flow rate of 1 mL/min. The peak of the GA present in the extract was identified by comparing the retention time (Rt) of the reference standard at 3.00 min with no interference at 280 nm and the peak of EA was identified at 5.5 min with no interference at 254 nm. In both the cases good correlations were found between concentrations and the peak area, with the correlation coefficient of more than 0.99. Figures 1 and 2 represents the chromatograms obtained from HPLC analyses of the extract, standard GA and EA. The content of GA and EA in the test sample was found to be 0.41% (w/w) and 0.16% (w/w) respectively.

Figure 1: A. HPLC chromatogram of standard GA and B. HPLC chromatogram of T. arjuna hydro alcoholic extract.

Figure 2: A. HPLC chromatogram of standard EA and B. HPLC chromatogram of T. arjuna hydro alcoholic extract.
Determination of CYP450 concentration and percentage inhibition through CYP450-CO Complex assay
Cytochrome inhibition assay through cytochrome P450-CO complex method in presence of specific CYP inhibitor is the first approach to study the inhibitory potential. Concentration-dependent inhibition of CYP450 was shown that test sample and its constituents had much less potential to inhibit the CYP enzymes. The concentration of protein in isolated rat liver microsome was found to be 7.7 mg/mL. Cytochrome P450-CO assay was used to assess the inhibitory potential of T. arjuna. Ethanol and DMSO solution of the extract, GA and EA indicated a concentration reliant inhibition of CYP450 (Figure 3A and 3B). The CYP450 concentration of the diluted liver microsome was calculated to be about 0.304 nmol/mg protein. Results shown that plant extract, GA and EA exhibited significantly less inhibition than Ketoconazole and Quinidine (Figure 3C). Two different solvents were used to evaluate the CYP450 inhibition, the results revealed minor variation; this confirms that the solvent effect was minimized. T. arjuna extract (31.02 ± 2.24%), GA (28.14 ± 2.45%) and EA (18.00 ± 1.16%) in DMSO showed slight highest percentage of inhibition. An organic solvent such as DMSO and ethanol strongly affect the metabolism at 5% concentration level [9]. The variation in inhibition was detected with all the DMSO solubilized extract, when compared with the ethanol solubilized extracts. But, to confirm the possible inhibition by the test samples, suitable solvent controls were used in the study and the percentage inhibition was calculated after counteracting the solvent effect. So, it was confirmed that the concentrations of DMSO and ethanol used in this study did not interfere in the CYP450 interactions.

Figure 3: A and B. Concentration dependent inhibition of the T. arjuna extract, GA and EA in DMSO and Ethanol. C: Percentage inhibition of T. arjuna, GA and EA versus positive control Ketoconazole; Values are expressed in mean ± SEM; n=3; ANOVA followed by Dunnett's multiple comparison Test. Level of significance *p<0.05, **p<0.01 and ***p<0.001.
Determination of IC50 by high throughput fluorometric assay
Fluorometric assays helps to ensure the drug interaction potential of test compound through the important CYP isoforms (CYP3A4, 2D6). Inhibitor potency was quantified by determination of IC50 values. T. arjuna extract, GA and EA were assayed between concentrations ranging from 1.5 to 25 μg/mL. All tests were assayed in triplicate, and IC50 values were calculated using endpoint mode (Table 2). Concentration reliant percentage inhibitions of the test compound on CYP3A4 and CYP2D6 were observed (Figure 4). The study results shown that the T. arjuna extract and the constituents had less inhibition potential on the CYP3A4 and CYP2D6 compared to their respective positive controls. IC50 values for positive inhibitors were in good agreement to previous studies [19]. T. arjuna in DMSO exhibited a better activity against CYP3A4 and CYP2D6 but IC50 value (48.06 ± 1.14-57.89 ± 2.15 μg/ mL) seemed high when compared to the respective positive controls (07.20 ± 0.56-06.28 ± 1.76 μg/mL). Results indicated that the test extract have higher inhibition potential comparing to their individual bioactive compounds. The higher enzyme inhibition potential by the extracts may be related to the synergistic effects due to presence of other constituents in the extract.
| Test sample | Solvent | IC50(µg/ml) (CYP3A4) |
IC50 (µg/ml) (CYP2D6) |
|---|---|---|---|
| T.arjuna | DMSO | 48.06 ± 1.14a | 53.66 ± 1.36b |
| Ethanol | 53.36 ± 2.45a | 57.89 ± 2.15b | |
| GA | DMSO | 66.54 ± 1.04a | 78.46 ± 1.32b |
| Ethanol | 72.13 ± 2.67a | 83.84 ± 1.06b | |
| EA | DMSO | 69.47 ± 1.18a | 97.34 ± 1.55b |
| Ethanol | 74.32 ± 2.08a | 102.69 ± 2.87b | |
| Ketoconazole | DMSO | 07.20 ± 0.56 | - |
| Ethanol | 07.74 ± 1.32 | - | |
| Quinidine | DMSO | - | 05.84 ± 0.68 |
| Ethanol | - | 06.28 ± 1.76 |
Values are expressed in mean ± SEM, aP<0.001, bP<0.001 versus positive control ketoconazole and quinidine respectively. Abbreviations: DMSO: dimethyl sulfoxide; GA: gallic acid; EA: ellagic acid
Table 2: IC50 (μg/mL) value of T. arjuna extract, GA and EA on CYP3A4 and CYP2D6.

Figure 4: Concentration dependent inhibitory effect of T. arjuna extract and bioactive compounds on CYP3A4 in DMSO (A) and Ethanol (B) versus Ketoconazole, CYP2D6 in DMSO (C) and Ethanol (D) versus Quinidine.
Docking studies of active constituents and controls within CYP3A4 and CYP2D6 molecule active site
Molecular docking analysis on the interactions of GA and EA to CYP3A4 and CYP2D6 were performed to confirm the drug binding conformation. GA and EA had docking score of -8.55 and -8.36 kcal/mol with CYP3A4 respectively, which is lower than that of the Ketoconazole-CYP3A4 complex (-9.10 kcal/mol) (Figure 5E). GA has been interacted via van der Waals with PHE 302, ASN 451, GLY 444 (Figure 5A) and EA was interacted with THR 310, ALA 305, PRO 434 and ALA 370 respectively with CYP3A4 (Figure 5B). With CYP2D6, GA and EA had docking score of -7.44 and -8.17 kcal/mol respectively, which is less than that of the Quinidine-CYP2D6 complex (-10.48 kcal/mol) (Figure 5F). GA also interacted via van der Waals with PRO 435, SER 437, HIS 376, MET 374, ARG 101 (Figure 5C) and EA was interacted with SER 473, PRO 435, MET 374, HIS 376, ARG 441, and ARG 101 AND ALA 305 respectively with CYP2D6 (Figure 5D). It was found that all the ligands were involved in hydrogen bonding with the binding site residues of CYP450. Overall molecular docking studies concluded that GA and EA have less glide score and glide Energy with both CYP3A4 and CYP2D6 when compared to respective positive controls energy and score. Molecular docking results indicated that GA and EA has weaker inhibition activity of the CYP3A4 and CYP2D6. Therefore the in-silico prediction was in agreement with the results obtained from the CYP3A4 and CYP2D6 inhibition assays.

Figure 5: Predicted binding sites poses of GA (A) and EA (B) within the active site of CYP3A4 molecule by docking, GA (C) and EA (D) within the active site of CYP2D6, Ketoconazole (E) within the active site of CYP3A4 and Quinidine (F) within the active site of CYP2D6.
Cytochrome P450 enzymes have been of particular interest in the field of drug discovery for numerous reasons including the involvement of these enzymes in the metabolism of over 95% of the drugs on the market and the potential of drug-drug interaction through metabolism. Drug interactions are not limited to synthetic drugs, herbal products may also act as triggers for changes in the CYP activity; for example CYPmodulating effects are described for echinacea, grape fruit juice or St. John's Wort [20,21]. Administration of herbs and allopathic medicine may increase the chance of herb-drug interaction, which results in an increase of the plasma drug concentration and produces toxicity. To avoid or minimize toxic drug-herb interactions, it is important to identify drugs that can interact with herbs using proper In vitro, in silico and in vivo models in the early stages of drug development. Through this study an approach was made to evaluate the interaction potential of T. arjuna with conventional pharmaceuticals.
A number of In vitro systems can be used to investigate herb-CYP interactions (e.g., liver microsomes, precision-cut liver slices, cultured hepatocytes, and cDNA-expressed enzymes) [22-26]. We have adopted a high-throughput approach to screen the inhibitory effect of test substances on two major drug-metabolizing CYP enzymes. High throughput screening assays may represent a useful strategy for the study of herb-CYP interactions. They are capable of handling the great number of herbal constituents, and have the ability to provide In vitro inhibition data as a criterion for monitoring herb-drug metabolic interactions involving human CYP enzymes. Cytochrome inhibition assay was performed through cytochrome P450-CO complex method in presence of specific CYP inhibitor. Concentration-dependent inhibition of CYP450 was shown that test sample and its constituents have much less potential to inhibit the CYP enzymes.
In silico approaches represent a useful tool for the study of herb- CYP interactions as demonstrated by our study. Our established pharmacophore model could readily distinguish the most potent inhibitor if CYP3A4 and 2D6. Thus, this model could be used as a high throughput-screening tool to identify natural constituents of herbal preparations that inhibit CYP3A4 and 2D6, before undertaking In vitro determinations [27]. This will help avoid co-administration of drugs that are extensively metabolized by CYP3A4 and 2D6 with herbal products that showed potent inhibitory effects on these enzymes.
However, the results shown that the T. arjuna extract and its bioactive molecules studied in this study inhibits the CYP3A4 and CYP2D6 with an IC50 values of >0.103 mg/mL. In contrast, a relatively strong inhibition was observed with known inhibitors such as quinidine and ketoconazole with an IC50 value of <0.0078 mg/mL. From these findings, it was observed that the test substances have very weak inhibitory potential on CYP450 and its important isoforms (CYP3A4, CYP2D6). The order of inhibitory potential of the test substances through In vitro results were identified as TA>GA>EA. The extract and its bioactive molecules tested in the present study had very weak interaction with other drug biotransformation but the observed inhibitory effect on metabolizing enzymes can be highly variable as the constituent content can differ with plant species, source, environment, and processing and storage conditions.
However, herb-drug interactions are difficult to characterize and resolve, because of the lack of comprehensive federal regulations regarding safety, efficacy, and manufacturing standards for herbal medicines. It has been recommended that herbs are appropriately labelled to alert consumers to possible interactions with other concomitantly used drugs and to recommend a consultation with their general practitioners, pharmacists, and/or other medical carers. It is time to consider herbs not as alternative medicine based on tradition and experience, but as Phytotherapy, an integrated part of medical treatment [28]. Regulations with regard to safety (e.g., herb-drug interactions), quality and efficacy of herbs would be highly desirable. Thus, monitoring of adverse events when herbal medicines are co-administered with drugs can be systematically carried out and potential herb-drug interactions be identified. This would enable more accurate product labelling and a body of useful information on potential herb-drug interactions to medical professionals.
In conclusion, our study highlighted the inhibitory potency of T. arjuna and its standardized therapeutic bioactive compounds on CYP450 and its significant isoforms. Even though results indicated that test extracts and bioactive molecules had weak interaction potential, time interval should be considered based on elimination half-life of therapeutic drugs when T. arjuna containing herbal formulation administered. In vitro and in silico evaluation of Herb-CYP450 interactions can be incorporated to find out the interaction inducing molecules in the early stages of drug development.
We would like to thank Department of Science and Technology, Government of India (Grant number: VI-D&P/372/10-11/TDT) for their financial assistance and support. We thank Dr. M. Muthu Tamizh (CCRS, Chennai) and Dr. RC Satish Kumar (IIISM, SRM University) for helpful discussion.
The authors declare that there are no conflicts of interest.
