Review Article - (2015) Volume 4, Issue 2
The mitochondrion is the only eukaryotic organelle with its semi-autonomously controlled genome. The minuscule nature of this genome does not undermine the importance of its products in cellular functions. This double-stranded circular genome sustains numerous mutations that have been useful to molecular anthropologists in phylogenetic studies. Some of the mutations sustained by this genome are rather pathogenic, being involved in a wide spectrum of human diseases. In recent times, the importance of mitochondrial mutations in cancer is being established. However, there are still “dark or enigmatic” spots in our knowledge in regard to making an attempt to explain various mitochondrial pathologies. The recent uncovering of novel and, possibly coding fusion genes, as well as noncoding transcripts should usher in a new era of knowledge about this genome. The importance of these novel transcripts in diagnostics and pharmaceutical targeting in various diseases awaits further discovery and validation.
The eukaryotic cell established a symbiotic relationship with the mitochondrion, a prokaryotic cell, in order to enhance energy production essential for its normal cellular functions. This relationship has been very successful as evidenced in the inter-relationships between these two primitive cells. Despite this interdependency, there is a level of autonomy in their genetic regulation. Thus, the mitochondrion has its own genome that encodes a minuscule, albeit important, complement of its functional proteins, as well as its transfer and ribosomal RNAs required for the translation of its mRNAs. Expectedly, therefore, genomic aberrations in nuclear genes that encode proteins required for mitochondrial functions, as well as mitochondrial genome (mtgenome) changes can all affect the functions of this organelle. Hence, some diseases, including progressive external ophthalmoplegia (PEO), Pearson syndrome (PS), Kearns-Sayre syndrome (KSS), mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), myoclonic epilepsy with ragged red fibers (MERRF), Leber's hereditary optic neuropathy (LHON) and neuropathy, ataxia and retinitis pigmentosa (NARP) syndromes are attributed to mitochondrial genome alterations. A catalogue of diseases due to mtgenome mutagenesis is provided in Table 1.
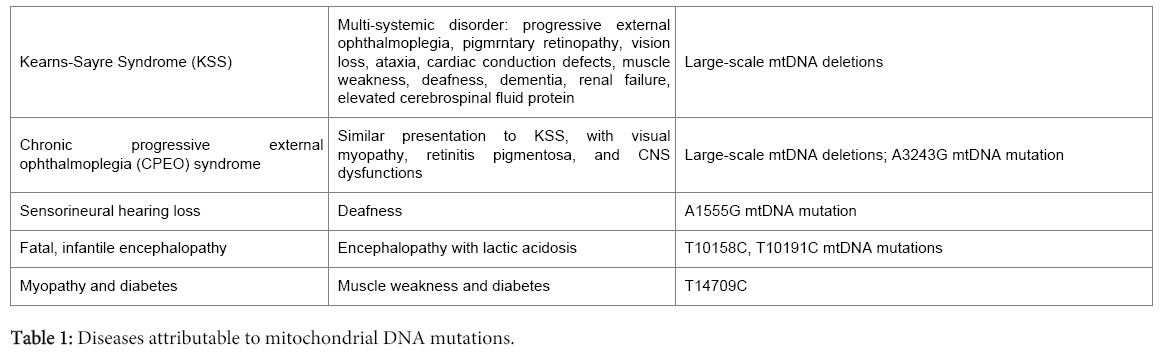
Table 1: Diseases attributable to mitochondrial DNA mutations.
While the “conventional or canonical” knowledge of mtgenome alterations in disease such as those alluded to above is grounded, recent evidence reveal a more complex mtgenome changes in various other diseases including cancer. At present, there are demonstrable evidences of mitochondrial transcription of noncoding RNAs involved in cancer. Moreover, we have established evidence of mitochondrial fusion transcripts expressed in cancer samples. These fusion genes demonstrate translational potential because of the presence of open reading frames (ORF). This synopsis is to stimulate our appetite for further inquiry into these molecules, and their potential roles in other human diseases apart from cancer.
Mitochondria are specialized eukaryotic organelles acquired several billion years ago by the eukaryotic cell in a symbiotic relationship with α-proteobacteria. This relationship led to functional specialization such that the mitochondrion became the site of most energy production required for normal cell physiology. In addition to being the only organelle with its own genome, the mitochondrion has several unique features worthy of a summary:
• The mtgenome is small and circular in nature, which contrasts with the large and linear nuclear genome.
• The mitochondrial genes are compactly organized without intervening introns as seen in the nuclear genome; hence there is no splicing of primary mitochondrial transcripts.
• It is primarily inherited in a uniparental fashion, being passed on from mother to offspring.
• The genomes are polyploidy, with each cell possessing hundreds to thousands of mtgenomes.
• As a genome with such high polyploidy, alterations in some genomes give rise to the concept of heteroplasmy, which refers to the co-existence of mutated and wild type genomes in the same organelle and/or cell.
• For mitochondrial disease to manifest, mutant copies must outweigh wild type genomes in proportion, and this is referred to as the threshold effect.
• This genome has a high mutation rate as a consequence of its residence in an organelle that generates most of the reactive oxygen species in the cell.
• The translation of mitochondrial mRNAs relies on mitochondrialspecific genetic code that is slightly different from the nuclear genetic code.
• Finally, mitochondria segregate in a stochastic fashion to daughter cells during cell division.
All these unique features have implications in mitochondrial diseases and genetic technologies such as cloning and production of stem cells.
Nass and Nass [1] first reported on the mitochondria having its own genetic material, and the Sanger’s group [2] sequenced this genome nearly two decades later. Structurally, the mtgenome is a compact prokaryotic-like double stranded genome composed of 16568 nucleotide base pairs (Figure 1). This genome encodes 37 molecules involved in its functions. These molecules are the 2 rRNAs and 22 tRNAs used for mitochondrial translation, and the 13 proteins that complement the nuclear encoded proteins involved in the electron transport chain and oxidative phosphorylation. Except for mitochondrially encoded NADH dehydrogenase 6 (MTND6), all the 13-mitochondrial polypeptides are encoded using the heavy or Hstrand. Additionally, the H-strand encodes 14 of the 22 tRNAs and the 2 rRNAs. Mitochondrially encoded NADH dehydrogenase 6 and the remaining 8 tRNAs are encoded by the light or L-strand. There are two noncoding mitochondrial regions. The first is a large segment referred to as the D-loop, which is composed of 1121 bp, and extends from nucleotide position (np) 16024 to np 576. This region houses regulatory elements for mtgenome transcription and translation. Within the L-strand origin of replication is another noncoding region composed of just 30 nucleotides (nts). The structural complexity of this genome belies what is being uncovered at the moment. It is my opinion that there is a lot yet to be revealed, and their role in normal physiology and pathophysiology will be illuminating.
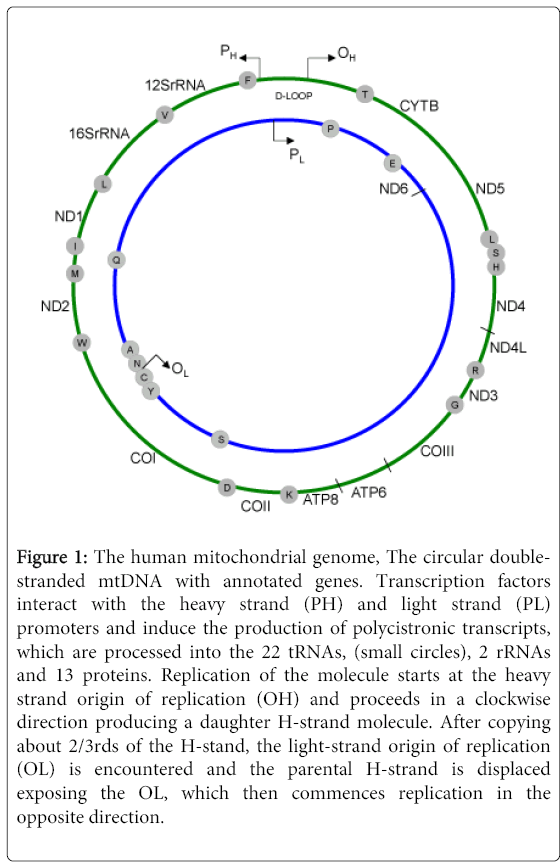
Figure 1: The human mitochondrial genome, The circular doublestranded mtDNA with annotated genes. Transcription factors interact with the heavy strand (PH) and light strand (PL) promoters and induce the production of polycistronic transcripts, which are processed into the 22 tRNAs, (small circles), 2 rRNAs and 13 proteins. Replication of the molecule starts at the heavy strand origin of replication (OH) and proceeds in a clockwise direction producing a daughter H-strand molecule. After copying about 2/3rds of the H-stand, the light-strand origin of replication (OL) is encountered and the parental H-strand is displaced exposing the OL, which then commences replication in the opposite direction.
The mtgenome encodes noncoding transcripts of biologic importance. Currently, a number of groups have demonstrated the presence of noncoding mitochondrial RNAs (ncmtRNA). Initially reported in the mouse, the human equivalent of the ncmtRNA was uncovered in 2007 [3]. This molecule is composed of an inverted repeat sequence that is covalently linked to the 5’ end of the mitochondrial 16SrRNA transcript. The human long noncoding RNAs are ~2374 nt long that is organized into a stem-loop structure composed of ~820 bp double stranded stem and a 40 nt loop. The stem is resistant to RNase A digestion. Putatively, this transcript complex with cytochrome c and endonuclease G in mitochondria, and its expression is associated with proliferating cells. Subsequent analysis indicated proliferating cells express both sense and antisense transcripts [4]. The antisense ncmtRNAs are two different inverted repeats linked to the 5’ ends of the antisense 16SrRNA transcribed from the L-strand.
The biological importance of these novel molecules has been demonstrated in a number of follow-up studies. Normal proliferating cells express both transcripts (sense and antisense). However, analyses of 15 different cancer cell lines and 273 cancer biopsies from 17 different cancers indicate the complete absence of the antisense transcript, with expression of only the sense ncmtRNA. This suggests the down regulated expression of the antisense transcript is a tumourspecific biomarker [4]. It has also been shown in human renal cells that these molecules translocate into the nucleus and associate with heterochromatin. In cancer cells, as expected, only the sense molecules form complexes with heterochromatin [5]. In a pilot study of urinary samples, both transcripts were not detected in exfoliated cells in urine from healthy individuals. However, urine samples from bladder cancer patients expressed the sense transcript in association with loss of the antisense noncoding transcripts, and this aberrant expression was able to discriminate samples from cancer patients and healthy control subjects [6]. The decreased expression of the antisense molecule appears to be a hallmark of cancer. Indeed, it is demonstrated that high-risk human papilloma virus oncoproteins modulate the expression of these ncmtRNAs. Human papilloma virus E2 oncoprotein represses the expression of the antisense transcript, while E6 and E7 induce the expression of yet another sense transcript referred to as SncmtRNA-2. This novel transcript share 3’ sequence homology with SncmtRNA-1, but has a diverse 5’ sequence structure [7].
Rackhman et al. uncovered three novel long noncoding RNA (lncRNA) of mitochondrial origin by close analysis of deepsequencing data [8]. Their presence was authenticated by Northern blotting and qRT-PCR analyses. These molecules are mainly encoded by the light strand of the mtgenome and are complementary to MTND5, MTND6 and MTCYTB genes. The expression levels of these lncRNAs were at 58%, 34% and 14% respectively to their traditionally known coding complement. These molecules are also partners of the nuclear-mitochondrial communication network, because nuclear encoded genes that regulate RNA processing are also involved in their processing. And mitochondrial RNase P protein 1 (MRPP1) has been identified to play a role in their processing. These molecules form intermolecular duplexes that resist RNase 1 digestion, suggestive of their possible functions in the regulation of their complementary coding mRNA. Putatively, the MTND6 lncRNA molecule protects the non-polyadenylated MTND6 coding molecule through this duplex formation. On the flip side, this duplex formation reduces the levels of translated products from MTND6, which is established to be less abundant compared to other mitochondrial proteins.
Further complexity of this genome is the recent finding of a novel class of abundant small noncoding RNAs, analogous to miRNA [9]. Referred to as mitosRNA, these molecules range in sizes from 20-40 nt long, with many composed of 30 nts. Their abnormal expression was associated with aberrant mitochondrial transcription suggestive of a regulatory role by mitosRNA. While these small ncmtRNAs resemble miRNA, it appears they are not necessary processed by DICER. The polymerases involved in their expression, as well as their processing molecular machinery are yet to be uncovered.
The mtgenome is studded with several repeat sequences that mediate mtDNA large-scale deletions. Indeed, the computermodelling estimate in 1989 by Schon [10], revealed a modest 331 repeats, consisting of 253-10 bp, 58-11 bp, 16-12 bp, 3-13 bp and 1-15 bp repeats. Using recent computational models, thousands of deletions from these repeats are predicted in the minuscule mtgenome based on all possible combinations of repeats. But note that not all deletions are flanked by repeats, indicating even a higher number of mtgenome deletions. It is established that the majority of the deletions are flanked by these repeats, which are disproportionately distributed in the genome. Extending clock-wisely from MTCYTB to MTCOI genes and flanked by OL and OH harbours numerous repeats compared to the rest of the genome. Hence a large number of deletions occur in this region, and for that reason it is appropriately referred to as the major deletion arc. But extending counter-clock-wisely from nucleotide position 547 in the D-loop to about np 5443 in the MTND2 gene is a region paucity of repeats, and hence is associated with fewer deletions. This region is the minor deletion arc of the mtgenome.
Mitochondrial deletions are subcategorized as class I, II or III based on the presence and/or nature of the flanking repeats. While the vast majority (60%) have flanking short homologous direct repeats (class I), and about a third of the deletions have imperfect repeats (class II), there are a few that do not show any evidence of repeats (class III). Following a large-scale deletion, the part of the mitochondrial DNA removed from the genome is referred to as the deleted region, while the remaining molecule that re-circularizes and maintains replication origins, and hence capable of replication is referred to as deleted mtDNA. This deleted mtgenome can exist as a monomer, whereby it simply re-circularizes. In other instances, more than one deleted molecule can recombine to form deletion dimers or higher order multimers. Rarely, one or more deleted molecule(s) may recombine with a wild type-undeleted molecule to form partial duplications or triplications. Other rare rearrangements including inversions, simple insertions involving small number of nucleotides (up to 300) and complex forms involving kilo base insertions generating molecules of up to 20 kb, have been reported. The simple monomeric forms constitute the majority of these molecules (Figure 2).
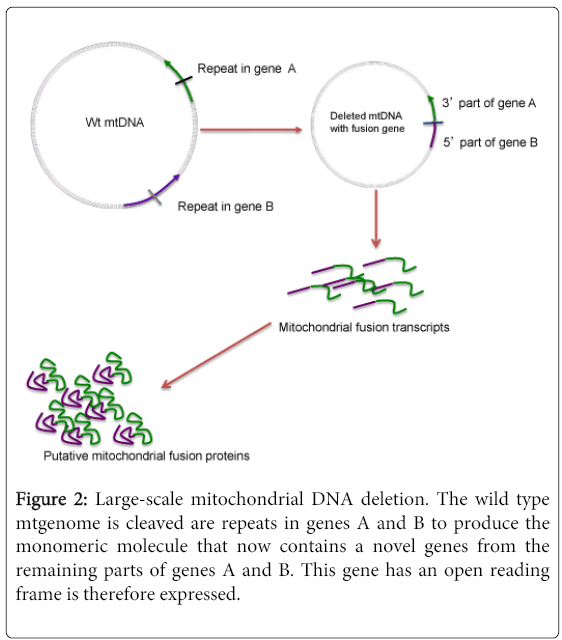
Figure 2: Large-scale mitochondrial DNA deletion. The wild type mtgenome is cleaved are repeats in genes A and B to produce the monomeric molecule that now contains a novel genes from the remaining parts of genes A and B. This gene has an open reading frame is therefore expressed.
The first reported large-scale deletions in the mtgenome were in muscle mitochondria from patients with mitochondrial myopathies [11]. Shortly following this initial report, it was demonstrated that these deletions were present in muscle mitochondria from patients with KSS [12], and that they affected COX activity. These deletions characterize the PEO and KSS spectrum, and they vary in sizes from 1.3 to as large as 7.6 kb deletions. They occur at various heteroplasmic proportions, but the levels do not appear to reflect on disease presentation or severity. A 4977 bp mtgenome deletion in the major deletion arc, referred to as the common deletion has been extensively investigated in patients with KSS and PEO. The deletion is defined by 13-bp repeat breakpoints at positions 8470-8482 in the MTATPase gene and downstream at position 13447-13459 in the MTND5 gene (Figure 3). This deletion thus removes parts of MTATPase and the MTND5 genes, and four entire intervening genes, namely MTCOIII, MTND3, MTND4L and MTND4.
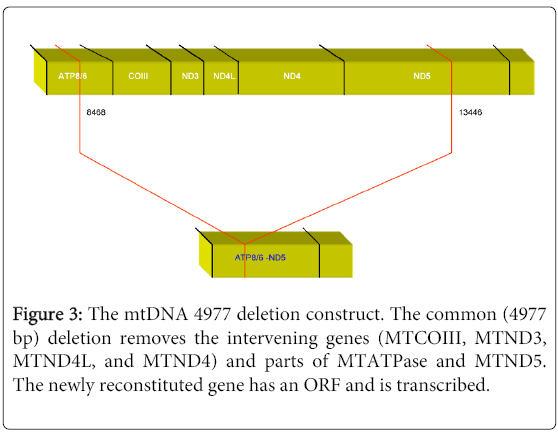
Figure 3: The mtDNA 4977 deletion construct. The common (4977 bp) deletion removes the intervening genes (MTCOIII, MTND3, MTND4L, and MTND4) and parts of MTATPase and MTND5. The newly reconstituted gene has an ORF and is transcribed.
After deletions in the mtgenome, the rejoining of the ends of the deleted molecule brings parts of two genes together to create a new sequence. The deleted upstream and downstream genes retain their initiation and termination sequences respectively. Extensive analyses indicate almost all these genes connect by generating novel genes with open reading frames, suggestive of their translation (Figure 4).

Figure 4: DNA and protein sequences from the fusion gene formed following a large-scale deletion associated with Wolfram syndrome. Note the output by the Open Reading Frame finder.
Many investigators have demonstrated the transcription of these fusion genes in various organisms and disease states. Deleted mitochondrial fusion gene expression has been reported in soybeans [13], fly [14], and in patients with KSS/PEO [15,16]. With reference to cancer, Horton and coworkers [17] first reported on differential expression of mitochondrial fusion transcript, and recently by our group [17,18, Dakubo unpublished]. Using Oligo-dT primers for recovery of these transcripts, we demonstrated the fact that in addition to having an ORF, the transcripts from these genes are indeed polyadenylated. Their role in carcinogenesis and other disease pathogenesis is obviously suggestive by our subsequent work. Analyses of multiple primary tumours, body fluids and cell lines reveal differential levels of expression of these transcripts between samples from cancer patients and healthy control subjects. While the functional role of these molecules is currently unappreciated, there is a need to unravel the importance of their role in the diagnosis, prognosis and possible predictive utility in various diseases.
It is counterintuitive as to why a cell will transcribe a gene with an ORF, and post-transcriptionally process the mRNA (at least by polyadenylation) and not complete the process of translating this molecule into a functional protein. A number of investigators have thus sort to detect these fusion proteins, but with some difficult [15,19]. However, the idea had persisted because not all the mtgenomes in a particular mitochondrion will be deleted, and hence the possibility of intraorganellar complementation should enable sufficient rRNAs and tRNAs necessary for translation of the fusion genes to occur. But this process somehow will be dictated by the extent of the heteroplasmic proportions of mutant genomes. Thus in 1991, Hayashi et al. [18] demonstrated for the first time the translation of mitochondrial fusion genes. Using cells with various heteroplasmic levels of deleted genomes, they detected a narrow window whereby the translated fusion protein was visibly observed. This fusion polypeptide was evident in cells with 55-61% of deleted genomes. Translation in cells with > 60% (what Hayashi referred to as a “reflection point”) mutant molecules decreased. Putatively, when mutant mtgenomes above the “reflection point” occur, competition for mitochondrial tRNAs causes global decrease in translation of both wild type and mutant genomes.
These enigmatic molecules uncovered so far are likely the tip of the iceberg. Transcription of molecules from the light strand of the mtgenome is of particular interest because it had been known to only transcribe ND6 and the remaining 8 tRNAs and nothing else. It is likely the entire light strand is transcribed and processed into these noncoding transcripts. The functions of these molecules and how they are regulated will be of interest to know. Indeed, the demonstration of mitochondrial fusion proteins, and their functions, especially in disease states should add to our expanding knowledge of the molecular pathogenesis of various diseases. For instance a computational analysis reveals the possible subcellular localization of the common deletion (Figure 5). What exactly is the function of this deletion in the cell?
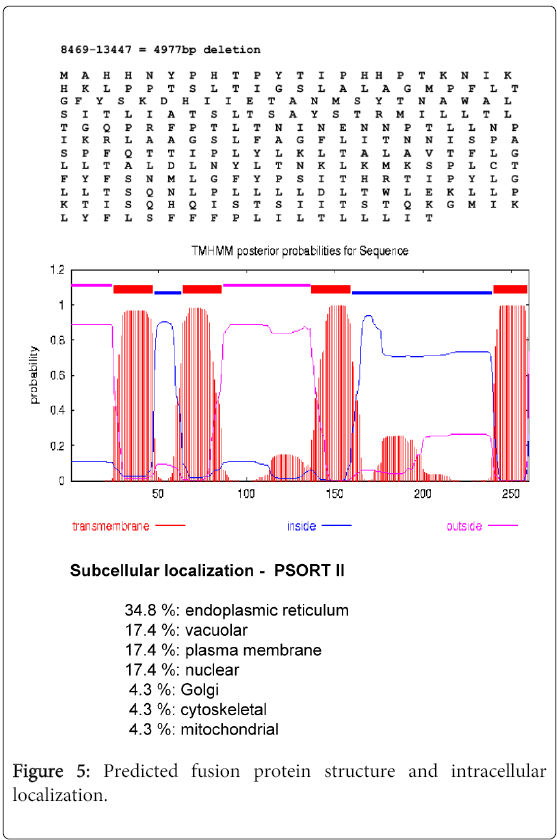
Figure 5: Predicted fusion protein structure and intracellular localization.
