Research Article - (2013) Volume 2, Issue 3
The most severe cases of autism are diagnosed by extreme social dysfunction and other behavioral abnormalities. A number of genetic studies have been conducted to correlate behavioral phenotypes to genetic dysfunctions, but no “autism gene” has yet been discovered. In addition, environmental factors have been found to influence the development of autistic traits with high probability. This review will examine the role of a shortened period of neuroplasticity as a unifying feature of the autistic phenotype. The neuroplastic period of interest normally extends into adolescence, allowing for neural integration and the development of language and social skills. Early closure of this period may result in a shortened period of development, forcing the brain to rely on underdeveloped structures.
Keywords: Autism spectrum disorder; Critical period; Plasticity; Language acquisition; Development
ASD: Autism Spectrum Disorder
Autism spectrum disorder (ASD) has recently been redefined in the Diagnostic and Statistical Manual of Mental Disorders-5. Symptoms required for diagnosis of autism and related diseases are being reclassified under the blanket term ASD. The reclassification will encompass diseases such as Asperger’s disorder under the ASD umbrella while removing diagnostic requirements associated with age of onset of symptoms. While these changes may over-generalize a wide spectrum of disorders, it simplifies the descriptions of social and behavioral deficits required for diagnosis. These include persistent deficits that are not a result of general developmental delay and include abnormal or reduced socio-emotional reciprocity, poor use of nonverbal communication, difficulty with age-appropriate relationships, repetitive speech use or movements, rituals or patterns of verbal or nonverbal behavior, unusual focus on objects or strange interests, and abnormal reaction to sensory input including fascination with light or spinning objects. Symptoms must be present in early childhood even if diagnosis is made later, and there must be a general impairment of daily functioning as a result of these symptoms [1,2].
ASD has been tied to single-point mutations, copy number variation, and polygenic mechanisms. There are high correlation rates from twin studies suggesting that many mutations increase the risk of ASD, but no single mutation causes ASD in 100% of cases, suggesting complex interactions influenced by both genes and the environment [3]. Pharmacological treatment has shown some benefit for treating ASD symptoms but there has been no effective cure found [4]. Diagnosis has also been complicated by a variety of physiological dysfunctions that have been associated with ASD such as in the gastrointestinal and immune systems, as well as the stereotypical effects on the brain [5].
Some of the better-known diseases that share genetic or physiological traits with ASD include schizophrenia, bipolar disorder, Fragile X syndrome, and ADHD [6-9]. The interplay of multiple genes in these disorders and the variety of outcomes suggests the interruption of a complex and vital network required for normal development. The symptoms of these diseases can manifest in a wide spectrum from mild to severe, complicating the search for an ultimate cause. Functional and structural defects are often used in genome studies, but the range and variety in phenotypic presentation of behaviors is difficult to incorporate as a factor due to complications in obtaining data. The other barrier to analyzing the spectrum of behaviors is the separation of information into different academic communities. Behavioral data is typically found in psychology literature and often with small sample sizes; while physiological, structural, and molecular data are typically relegated to separate scientific fields. In these cases, data from several communities and techniques may be required to observe a pattern.
An additional confounding factor in neural dysfunctions is the role of time. As the individual develops, symptoms change, manifest, or even fade. This change over time is critical for understanding both the problem and the cause of ASD. Some of the more interesting observations are made with this consideration, such as the symptom reducing effect of exercise, diet, or early behavioral intervention on autistic symptoms over the long term [10-12]. The focus on time, particularly into adolescence, has been studied in the disease state of ASD, but there has been no attempt to find a normal-functioning correlate to the autistic phenotype.
One of the main deficits in those with ASD is the lack of proper attribution of feelings to others, a sort of mindreading referred to as Theory of Mind [13,14]. The ability to accurately attribute mental states for those with ASD, further extends into failing to understand even their own mental states [15]. There is neural integration between the systems supporting visual processing, social cues, and Theory of Mind systems [16], and imaging studies depict the change in connectivity between these systems over time in normal subjects [17], suggesting activity-dependent growth interruption may lead to symptoms of ASD.
Activity-dependent growth interruption is supported by the Critical Period Hypothesis that proposes the existence of a normally occurring window of opportunity for the development of language and other skills requiring higher cognition, with a limited timeframe closing sometime in adolescence [18]. Critical periods generally refer to times during which the brain is sensitive to experience-dependent growth and refinement, when neural circuits are custom tailored to the individual beyond what is dictated by the genome, as influenced by the environment [19].
The Critical Period Hypothesis is supported by observations of survivors of various forms of depravation. For example, ‘feral’ children are individuals who were raised in isolation, with wild animals, or under conditions of extreme neglect. They exhibit a variety of mental deficits, and if behavioral therapy is attempted too late there is a roadblock at the acquisition of higher syntactical structures, suggesting the critical period has closed [20,21]. Feral children, even after considerable rehabilitation, exhibit many behavioral symptoms similar to ASD including impaired language skills, social deficits, and behavioral abnormalities [21-23]. In both ASD and in cases of extreme neglect or isolation, there has been some success with intense behavioral therapy when administered as early as possible. To take best advantage of this window of opportunity, recommendation for behavioral intervention in ASD is as young as 2-3 years [24].
The most interesting difference between ASD and cases of feral children is that similar phenotypes result from two very different causes. ASD is generally considered genetic, while feral children are a product of environment. Both types of deficit have had some success therapeutically, with the therapeutic window occurring during the critical period in childhood when the brain is more plastic. The closing of the critical period signals a significant reduction in plasticity, as neural networks, structures, and connections become consolidated [19].
Critical periods are times when experience has a strong impact on brain development [25,26]. In the visual cortex, for example, there is a limited window for the development of stereoscopic vision. If one or both eyes are deprived of normal vision during this time, vision will be permanently impaired. This critical period coincides with rapid synapse production in the visual cortex which is thought to accommodate the development of normal vision [26,27]. There are also periods for filial imprinting [28,29], speech sound recognition [30-32], and social and emotional behavior [29,33-35]. These critical periods rely on proper functioning of neural circuits that process experience [25].
Plasticity is the ability of neurons or the networks they comprise to change in response to these experiences. During periods of plasticity, the brain is particularly resilient, and can recover easily from some injuries that cause permanent damage in adults [36]. Plasticity also describes the ability to develop new functions through new axonal outgrowths, synapse elimination, and synapse consolidation [25]. Most axonal outgrowth begins at birth and ends at age 2-3 [37,38]. Dendritic growth extends slightly beyond that to year 4, and individual variation in dendritic trees is observed after age 5. The variation in dendritic trees is hypothesized to be the effect of experience and environment [39]. Some parts of the brain such as the frontal cortex continue growing until age 12-13 [40], still within this window of plasticity. A sequence of sensitive periods has been described in youth, where one period must close before the next may open; for example, if computation and consolidation are separate, the computational network receiving information must become reliable before consolidation can occur. Premature consolidation or consolidation after deprived or inadequate environments will therefore result in abnormal neural circuit connectivity in adulthood [25]. This abnormal connectivity provides a rationale for the difficulty in learning new skills, such as first language acquisition in those with insufficient language exposure during childhood.
The processes involved in synaptic plasticity rely on a variety of structural and signaling molecules [25], and plasticity is controlled by a signaling network associated with molecules regulated by neuronal activity. The synapse responds to neuronal activity and in turn triggers alterations in RNA translation, signaling for regular synapse function and control. These are the experience-dependent pathways necessary for the acquisition of new skills [41-43]. The behavior of synapses at different regions in the brain is used as a measure of plasticity [41,44,45]. Excitatory/inhibitory balance at the synapse is even suggested to be the controller of plasticity [26]. Structurally, the synapse is also reliant on the extracellular matrix and cellular adhesion molecules, both of which have been tied to synaptic plasticity and mental disorders, when dysfunctional [46,47], adding to the immense number of molecules necessary for proper functioning of the neural network.
Synaptogenesis peaks at 2-4 years and continues steadily to 10-15 years, concluding with neurotransmitter maturation around the time of massive synaptic pruning and consolidation in adolescence. The frontal and parietal cortices continue growing through years 12-14, presumably accommodating learning processes and incorporation of new experiences. In addition, myelination continues to increase from 14-21 years, indicating improved isolation of different electrical signals [39]. These changes are part of the consolidation of neural networks and overall decrease in neural plasticity observed during adolescence.
In addition to synaptic pruning and consolidation, typical neural changes during adolescence include hormonal changes [48], limbic system maturation [49], redistribution of fiber density across brain regions [50], increased myelination [39], decreased gray matter density [51] and new synapse formation [52]. These changes coincide with an overall decrease in synaptic plasticity. The limbic system matures just ahead of cognitive control mechanisms, and this unbalanced maturation is believed to be the cause of increased risk seeking behaviors observed in adolescence [53]. Hormonal influence during this time of sexual maturation also has long-lasting structural impact on neural plasticity and remodeling of cortical and limbic neural circuits [54-56]. Hormones such as estrogen and testosterone also have organizational and activational effects on the nervous system [57], and play a role in the addition of new cells in some areas of the brain such as the amygdala [58].
Synaptic pruning is so aggressive during this time that up to 50% of connections are removed in some brain regions [59]. One potential benefit of this massive pruning is energy efficiency, as synapses are energetically costly and the pruning process is conserved across species [60-62]. The speed and efficiency of information signaling across brain regions is also dramatically increased by a surge of myelination [63,64], resulting in the drastic increase in white matter, with a simultaneous decrease in gray matter [65]. This process helps to reconfigure brain activity, thinning out unnecessary connections, and allowing for better information processing [60].
During normal cognitive processes there are complex neural circuits being used across brain regions. The cerebellum provides timing control over the cerebral cortex and sensory systems. Networks between brain regions are supported by smaller networks within those regions and micro-scale networks between cell types. The complexity of this circuitry allows for an enormous number of potential mutations to interfere with its development. All of the molecular players in neural plasticity and development can impact the neural network and therefore the development of complex emotional and cognitive abilities [66]. A variety of psychiatric disorders, including ASD, have been tied to impairment of these connections [67-69].
There is a significant growth period when the brain is building these networks necessary for later cognitive development, and is highly plastic and susceptible to influence from variable social atmospheres and a host of environmental effects [70,71]. Some regions and developmental periods are more susceptible than others to damage [72]. The prefrontal cortex is responsible for higher cognitive processes like language and IQ [73], matures later than some areas, and behaviors relying on this area of the brain are more heritable than others, with heritability increasing with age [72]. This tells us that as the individual ages, the role of genes becomes increasingly important to cognitive abilities. This is paralleled by the decreasing impact of environment on shaping neural networks as the individual ages. The tradeoff between genes and environment over time suggests a window of opportunity for therapy and a supportive environment for those with debilitating genetic mutations.
Once this adolescent neural growth is complete it is more difficult for new experience to influence the brain as it grows. Several changes take place that lead to the loss of experience-based neural plasticity during young adulthood. While later experience elaborates and builds on what was created during youth, the foundation for this learning is essentially reliant on the scaffolding from early life experiences [74]. Because higher cognitive functions and the prefrontal cortex develop later, faulty mechanisms in early development can compound their impact on cognitive skills over time.
Diagnosis of ASD is usually around age 3 [75], and abnormalities are often observed by parents or experts by 12 months [76,77]. Age 3 is also the time when normal toddlers experience a peak in synapse density in areas of the brain linked to language development [70,78]. Studies suggest that infants acquire language through the modifications of constructs, refinement and fine-tuning of the brain space [79], while categorizing and mapping actions onto the brain [80]. This categorization and mapping is theorized to focus attention on items which are relevant, much in the same way that speech sound recognition narrows to the native tongue the child is exposed to [79]. It is possible that all sensory input is incorporated in this way; organizing and categorizing experience into informational items as the individual grows. In a child with ASD, if the neural networks are dysfunctional, this organizing feature would also be impaired and typical language impairments would be observed.
ASD is believed to be mainly of genetic origin, but the best genetic results are probabilistic correlations and not necessarily causal [81]. Four hundred to 1,000 loci have been implicated [82] but this has only compounded the problem of identifying a cause. The most specific and rare mutations seem to be the most helpful as they have higher correlations to ASD and show mechanistic connections across signaling networks [83]. For example, several mutations found in ASD are directly related to functions required by plasticity (Table 1). Many of these genes are known to be regulated by neural activity, including synaptic cell-adhesion molecules and others involved in the postsynaptic density in the dendritic spine head. Most of the genes listed in Table 1 are related to activity-dependent modifications and interact across complex pathways, as seen in Figure 1, making them necessary for learning and plasticity. These molecules associate with complex neural networks utilizing a vast array of molecules, any one of which has potential to cause a disorder if changed by mutation. This neural network model has already been proposed as an explanation for the wide diversity of mutated genes found in ASD, due to observed under connectivity across the neural network [83,84].
| Gene | Relevant function |
|---|---|
| CDH9/10 | Cell-to-cell adhesion, synapse development and maintenance [85] |
| CLTCL1 | Induced by neural activity [107,108] |
| FMR1 | Protein-synthesis-dependent plasticity of synapses [43,109] |
| HRAS | Activity-dependent development and plasticity [109-111] |
| MAP2K2 | Activity-dependent development and plasticity [109-111] |
| MECP2 | Possible genome-wide chromatin response to neuronal activity [112], experience-dependent synaptic remodeling [113] |
| NCKAP5L | Induced by neural activity [107,108] |
| NLGN3/4 | Synaptic adhesion [43] |
| NRXN1 | Synaptic adhesion [43] |
| PTEN | Protein-synthesis-dependent plasticity of synapses [43,109] |
| SEMA5A | Axon outgrowth guidance [114] |
| SHANK1/3 | Scaffolding in the postsynaptic density of excitatory synapses [115-117] |
| SHANK2 | Scaffolding in the postsynaptic density of excitatory synapses [115-117], synaptic plasticity in hippocampus [118,119] |
| SYNGAP1 | Activity-dependent development and plasticity [109-111] |
| TSC1/2 | Protein-synthesis-dependent plasticity of synapses [43,109] |
| UBE3A/B | Induced by neural activity [107,108], synaptic plasticity [120,121] |
| ZNF18 | Induced by neural activity [107,108] |
Table 1: Genes found altered in autism.
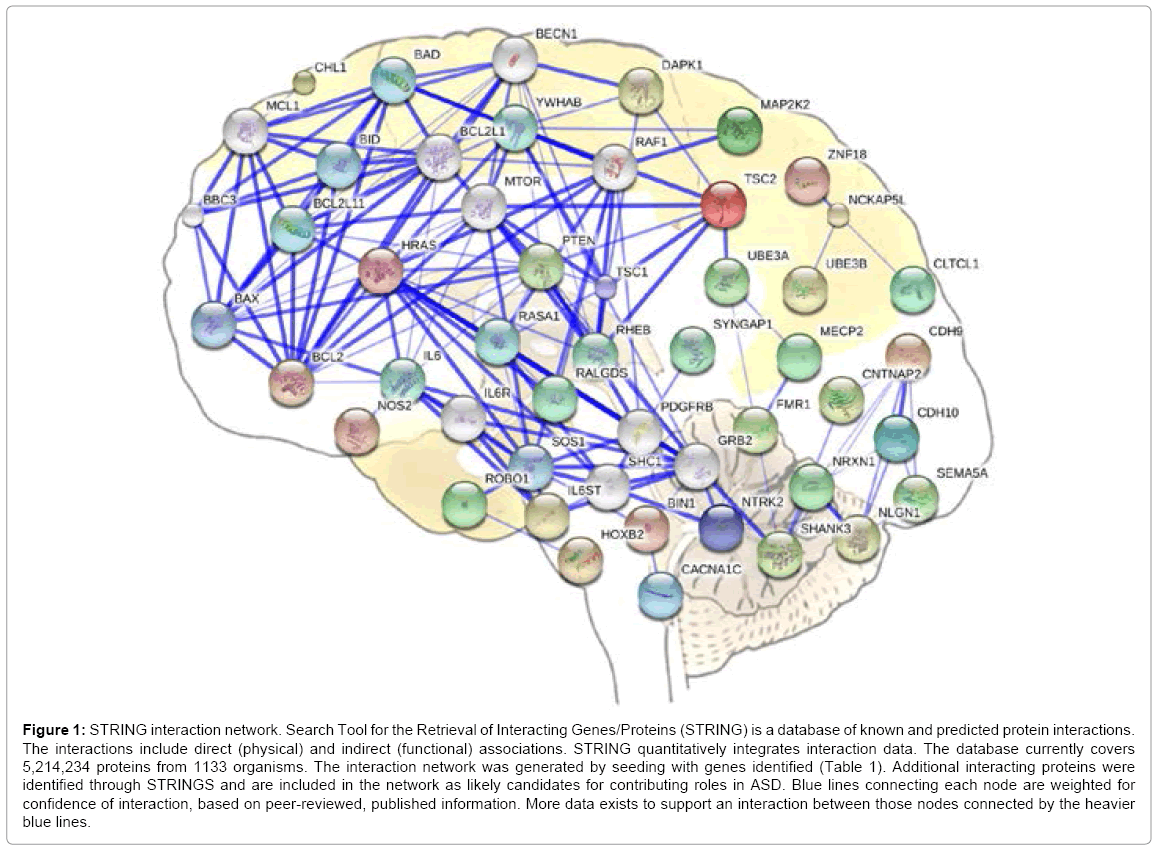
Figure 1: STRING interaction network. Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) is a database of known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations. STRING quantitatively integrates interaction data. The database currently covers 5,214,234 proteins from 1133 organisms. The interaction network was generated by seeding with genes identified (Table 1). Additional interacting proteins were identified through STRINGS and are included in the network as likely candidates for contributing roles in ASD. Blue lines connecting each node are weighted for confidence of interaction, based on peer-reviewed, published information. More data exists to support an interaction between those nodes connected by the heavier blue lines.
Recently, genome-wide studies have utilized diagnostic interviews for classifying the variety of phenotypes observed in ASD into categories for specialized gene analysis. Professional psychologists conduct interviews and perform the classification. This behavioral data is conducted in large sample sizes in the best cases, such as a recent study, which utilizes an array of statistical methods to achieve greater statistical power, such as the gene C80RFK32, with p-value of 3.44×10-8 [85]. However, most genetic studies do not agree on which genes are significant for producing the ASD phenotype. This leads to the general consensus that statistical methods need to remain under constant revision. Another possible explanation may be that genes need not hold statistical value across large sample sizes to tell us something important about the causal relationship. It may turn out that each gene found connected to ASD, even at low probability, is somehow included in the neural network required in its entirety for proper cognitive function. If this is the case, all pieces of this genetic puzzle, even statistically insignificant ones, may be necessary to see the full picture. The most interesting information might turn out to come from the least frequent mutations.
Treatments are speculated to require customization on the individual level due to the variability in genetic mutation across individuals. Mouse models provide hope for success with replacement therapy specific to the individual, but will require understanding of each individual’s genetic dysfunction at the synapse level [83]. Some of the most confounding aspects of autism have even been helped by these genome studies. For some time, the 4:1 ratio of male:female occurrence in ASD puzzled researchers, but recent studies now connect autism to the offset of hormone metabolism between genders and various genetic roles in development which may increase phenotypic presentation of symptoms in males. Females with the same mutations that give rise to ASD in males have stronger protection from the effects of these mutations [86].
Many potential triggers have been investigated for a causal role in ASD disorders, including sulfate levels [87], serotonin levels [88], pesticide exposure [89], maternal trauma [90], aluminum and acetaminophen [91], oxytocin [92], general diet [11], and mercury poisoning [93]. Most of these studies have demonstrated significant correlations; however, the fact that so many separate factors have a significant role in the development of a set of symptoms, suggests underlying factors may be at play. In addition, many of these causal factors have no apparent mechanistic relation to each other. The interactions are broad and simply too vast to posit a simple molecular explanation.
Brain volume in ASD increases rapidly from below normal at birth to 10-12% larger than normal at 2-4 years old. Volume growth ends around 2 years of age [70]. Increased cell packing and density in the limbic system and hippocampus in ASD in childhood is suggested to show early developmental curtailment [94], as these are areas that normally experience growth at a later time [49,53]. This confirms at least some abnormally timed growth patterns, and suggests the brain is maturing ahead of schedule.
General differences in ASD versus controls have been observed in volumes of white and grey matter distribution [95], control circuitry between brain areas [66,96], ion channel location and density [97], and general brain overgrowth [70]. When seeking for a cause, sometimes these differences are hypothesized to give rise to specific phenotypes. Causes of these structural abnormalities are then sought after, which in turn leads to related genes that influence the development of a given structure. But these genes may turn out to have little or no influence on the behavioral phenotype of ASD. This is one possible explanation for the low probability of cause-effect relationships in ASD-associated genes. A more successful approach may be to examine neural networks that include even the rarest of mutations and determine the causal relationship and associated physiological results afterwards. For example, brain-derived-neurotrophic factor (BDNF) is found in higher levels in ASD [5] and is shown to prematurely open critical periods when over expressed in mice [98,99]. Other mutated genes associated with ASD, such as those shown in Table 1, share the trait of being connected in some way to neural plasticity, and the mechanisms required for plasticity.
Brain overgrowth in ASD occurs in regions that normally mature during adolescence, and mutations occur in the network required for plasticity. This suggests an early closure of the critical period, and a loss of network consolidation that would normally occur in later life, had processes developed normally. This implies that the early environment is critical to individuals with ASD. The strength of environment as causal in these neural pathways is also apparent in the reports of success in early behavioral treatments for autistic symptoms [100]. 75-95% of cases now report the ability to develop useful speech if intervention is given before age 5 [101,102], demonstrating the shortened window into childhood of what normally continues into puberty.
The general consensus is that ASD is governed by an interaction between genes and the environment during critical developmental periods that are interrupted in a variety of possible ways [103]. Viewing ASD as a condition whereby premature closure of a critical period during adolescence occurs provides an explanation for the array of behavioral symptoms and abnormalities displayed in autism.
Early loss of plasticity deprives the autistic child of the developments that make sense out of sensations. Without the network consolidation that normally occurs in the maturing child, the autistic child remains a victim of the onslaught of stimuli we may imagine an infant receiving. Innumerable sights, sounds, and various other sensations, seemingly without coherence, will perpetually attack them from every angle. Luckily, with the passage of time and the assistance of therapy (and patience), these children are often able to sort out the bombardment of stimuli and acquire the ability to communicate. Sometimes with surprising wit and clarity [104].The genetic code has been described as a foundation or framework upon which our later abilities are built with influence from the environment [74]. Although there is significant plasticity loss in adolescence, there is still myelination and network remodeling throughout life [105,106] which at least suggests an individual with significant plasticity loss can still learn. The variety of genetic abnormalities that lead to ASD, the spectrum of symptoms and behaviors, and the range of therapeutic outcomes suggest that some of these mutations are more detrimental to the neural network than others. Many of these individuals most certainly retain the ability to learn (Table 1) [107-121]. The hope is that this opportunity may be recognized and taken advantage of by caregivers, leading to improved understanding and quality of life.
Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20RR016454 and P20GM103408, Biomolecular Research Center, and the Department of Biological Sciences (B451), Boise State University.
