Mini Review - (2015) Volume 4, Issue 1
Deletion of the short arm of chromosome 9 is associated with two distinct clinical prototypes. Small telomeric distal 9p deletions have been reported in patients 46,XY with gonadal dysgenesis, this region contains genes required in two copies for normal testis development. Recent studies have narrowed the interval 9p24.3-pter containing the putative autosomal testis-determining gene(s) known as domain DMRT. On the other hand, and depending on the extent of deletion of the short arm, the clinical characteristics of monosomy 9p syndrome may emerge. We present an infant female with complete 46,XY gonadal dysgenesis, who was examined for motor developmental retardation. In the karyotype a chromosomal deletion 9p24.1 was identified by cytogenetic and fluorescence in situ hybridization studies. No SRY deletion or mutation was detected. Ultrasonographic studies showed a normal uterus. Basal luteinizing hormone and follicle stimulating hormone values were high. The patient underwent gonadectomy at 3.2 years of age, and histologic analysis disclosed dysgenetic gonads with gonadoblastoma.
Keywords: Distal monosomy 9p chromosome; 9p partial deletion; Disorders of sexual development (DSD); 46,XY DSD; Gonadal dysgenesis; Fluorescence in situ hybridization (FISH); Gonadoblastoma; DMRT genes
Disorders of sex development (DSD) are congenital conditions which result in discordance between genetic, gonadal and phenotypic sex. In humans the primary event in sex determination depends on the presence and action of a master testis-determining gene, named sex determining region on the Y chromosome (SRY) which switches on the development of the testis direction. Other genes are also implicated in gonadal development, some of which are located in autosomic chromosomes, such as DMRT in 9p (OMIM 602424). Deletions and mutations of the SRY gene are present in 10-15% of DSD 46,XY female patients; deletions of 9p, which involve the DM domain, are associated with 46,XY DSD. Patients with this pathology also present mental retardation and typical features of monosomy 9p syndrome [1-4]. Here we report a patient with 46,XY DSD and partial deletion of the short arm of chromosome 9 that presents motor development delay, and normal female genitalia (Figure 1A).
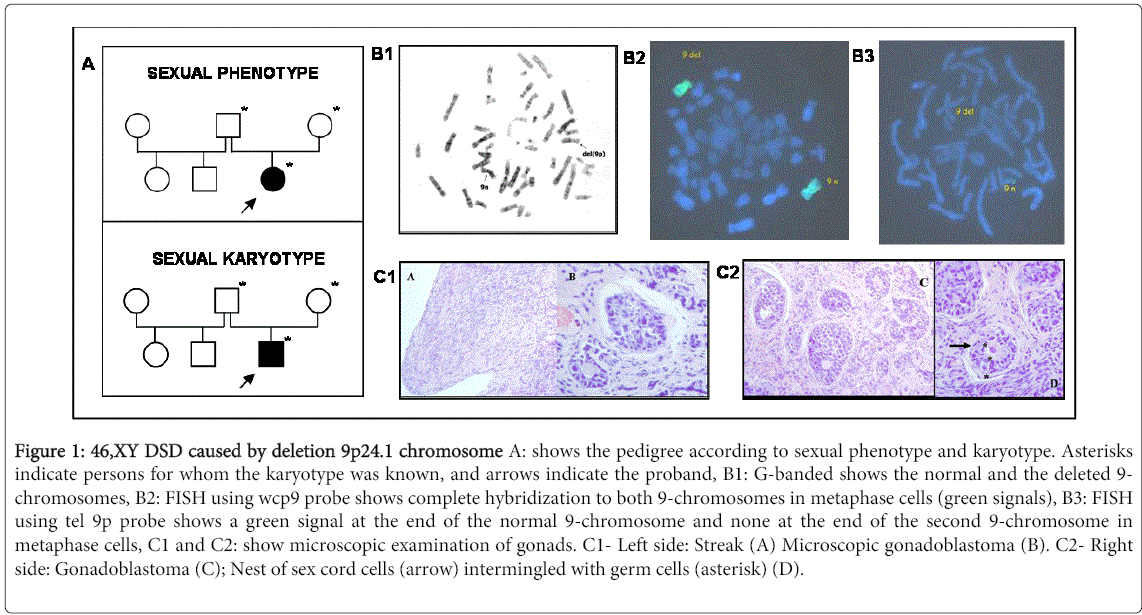
Figure 1: 46,XY DSD caused by deletion 9p24.1 chromosome A: shows the pedigree according to sexual phenotype and karyotype. Asterisks indicate persons for whom the karyotype was known, and arrows indicate the proband, B1: G-banded shows the normal and the deleted 9- chromosomes, B2: FISH using wcp9 probe shows complete hybridization to both 9-chromosomes in metaphase cells (green signals), B3: FISH using tel 9p probe shows a green signal at the end of the normal 9-chromosome and none at the end of the second 9-chromosome in metaphase cells, C1 and C2: show microscopic examination of gonads. C1- Left side: Streak (A) Microscopic gonadoblastoma (B). C2- Right side: Gonadoblastoma (C); Nest of sex cord cells (arrow) intermingled with germ cells (asterisk) (D).
Case report
The patient was born at 34 weeks of gestation from healthy nonconsaguineous parents (a 28-year-old mother and a 39-year-old father) after an uncomplicated pregnancy. Weight at birth was 2800 g. and the external genitalia had female appearance. The patient was referred to us at 2.3 years of age due to early motor development delay. Both weight and height were within the 25-50th centile; 12.100 Kg and 87 cm respectively. On physical examination mild facial dysmorphism was observed, including brachycephaly, narrow forehead, thin and dry hair, telecantus, depressed nasal bridge, big mouth, and micrognathia; left leg presented melanocytic spots. Ultrasonographic studies showed normal heart and kidneys and the presence of a well developed uterus, gonads were not visualized. Nuclear magnetic resonance imaging of the central nervous system showed abnormalities in white matter with underdeveloped frontal lobes. Results of ophthalmologic and otologic examination were normal. Endocrine laboratory studies revealed high LH (luteinizing hormone) and FSH (follicle stimulating hormone) basal levels (IFMA method). LH: 9.1 IU/L (normal range: until 0.3 IU/L) and FSH: 72.7 IU/L (normal range: 0.50-5.0 IU/L). Basal testosterone value was < 20 ng/dl (normal range: until 60 ng/dl, RIA method). The patient sat independently at 12 months of age and walked at 26 months of age. Her expressive language at 2.3 years of age was at a 16 month level, approximately. She showed persisting developmental delay and hypotonia, and required physical therapy.
Cytogenetic and molecular studies
PHA stimulated peripheral blood lymphocytes were cultured at 37ºC for 72 hours in McCoy's 5A medium plus 10% fetal calf serum and harvested for metaphase conventional preparations. Chromosome studies with GTG-banding were performed by the trypsin-Giemsa method.
Molecular cytogenetic by fluorescence in situ hybridization (FISH) studies were carried out on peripheral blood lymphocyte cultures using:
Whole 9-chromosome painting (wcp9) Spectrum green.
Telomeric 9p Spectrum green (tel 9p) which hybridize on 9pter.
All the probes were analysed and photographed using an epifluorescence microscope.
Molecular analysis performed on DNA extracted from peripheral blood lymphocytes included 1) polymerase chain reaction amplification of SRY, Y heterochromatin DYZ1 (Yqh), and DYZ3 (centromere Y) sequences. 2) single-strand conformation polymorphism analysis of SRY gene.
G-banding showed an abnormal 9-chromosome with 9p terminal deletion (Figure 1B1).
FISH with painting probe for whole 9, hybridized completely only in both 9-chromosomes in all cell lines (Figure 1-B2).
One small deletion at 9p24.1 until 9pter was shown when a telomeric probe 9p (tel 9p) was used. No translocations with others chromosomes were present (Figure 1-B3).
Karyotype was identified as: 46,XY,del(9)(p24.1).ish del(9)(wcp+,tel 9p-). Parents´s karyotypes were normal. No alterations were observed in the loci of SRY, DYZ1 and DYZ3.
The patient underwent gonadectomy at 3.2 years of age, due to the risk of developing a gonadal tumor. Bilateral streak gonads were found and histologic examination disclosed bilateral and microscopic gonadoblastoma (Figure 1 C1 and C2).
Deletions of various sizes on the short arm of chromosome 9 are associated with two distintic clinical entities. One small deletion to bands 9p24.3-9p telomere is associated with a non syndromic form of 46, XY DSD, being the DMRT (doublesex and mab-3 related transcription factor 1-3) the candidate genes for complete gonadal dysgenesis (OMIM 602424). There are three DMRT genes, namely DMRT1, DMRT3 and DMRT2 that encode proteins with a DM domain. DMRT1 is expressed only in the gonad and after sex determination in the fetal testis. The other entity is the 9pmalformation syndrome (OMIM 158170), which is assigned to bands 9p22.3-p24.1. Patients with larger deletions (distal or interstitial) leading to hemizygosity of many contiguous genes present a phenotype characterized by intellectual disability, delayed motor development and craniofacial dysmorphic features, such as trigonocephaly, long philtrum, and flat nasal bridge [5,6]. XY individuals with these conditions frequently present genital anomalies or testicular dysgenesis. The external genital phenotype ranges from complete female to male with hypospadias. The gonadal phenotype ranges from complete gonadal dysgenesis, or ovotestes, to cryptorchidism or hypospadias [7].
The patient in our study presented complete 46,XY gonadal dysgenesis caused by partial 9p monosomy. We performed FISH with a whole 9-chromosome painting and a telomere 9p probes to demonstrate a de novo deletion on 9p24.1-pter. The deletion was within the candidate region for gonadal dysgenesis [8,9]. She underwent gonadectomy which revealed the presence of gonadoblastoma.
Based on the literature reports and this study, we suggest that haplo insufficiency of a dosage-sensitive gene(s) in 9p24.3 might be responsible for the failure of testicular development and feminisation in XY patients with monosomy 9p [10]. The literature concerning this proposal is controversial [11,12]. Some of the 46,XY patients with similar monosomy 9p have been reported to present no abnormal genital phenotype or complete gonadal dysgenesis [13]. In these cases the phenotypic variability may not be only specific to haploinsufficiency of the 9p sex-determining gene(s). Quinonez et al. [14] propose a genomic detection of copy number variation (CNV) by array-comparative genomic hybridization in addition to monosomy 9p and DMRT haplo insufficiency, which may be needed for abnormal sex development. The fact that hemizygosity of many genes, or the unmasking of a recessive mutation on the normal chromosome 9 would be implicated in the variable gonadal phenotype might also be possible [7,15].
In summary, we present a new case of 9p- syndrome and 46,XY DSD, in which a gonadoblastoma was detected. More clinical and molecular data in similarly affected patients should hopefully elucidate the role of the 9p sex-determining genes in the fetalgonadogenesis.
The authors declare that they have no conflict of interests
We would like to thank Prof Susana Mancini for her permanent collaboration and Mr. Rodolfo De Bellis for the technical assistance.
