Research Article - (2014) Volume 2, Issue 6
Emodin (1) is the major bioactive compound of several herb species, which belongs to anthraquinone class of compound. As a part of our drug discovery program, large quantities of emodin (1) was isolated from the roots of Rheum emodi and a library of novel emodin derivatives 2–24 were prepared to evaluate their in-vitro antimalarial activity, among them, compound 17, 18 and 20 showed potent antimalarial activity against chloroquine resistant strain PfK1with the IC50 of 2.28, 2.49 and 2.48 μM respectively with a high safety index.
Keywords: Emodin derivatives; Antimalarial agents; Anthraquinones; Cytotoxicity; Rheum emodi
Anthraquinones are widely distributed secondary metabolites of plants (aloe, cascara sagrada, senna and rhubarb), microbes, lichens and insects, which possess various biological activities [1-3]. Emodin (1) an anthraquinone class of compound is the major bioactive compounds of numerous herb species such as Rheum, Polygonum (polygonaceae), Rhamnus (Rhamnaceae) and Cassieae (senna) [4-6] and possesses immunosuppressive, anticancer [7], anti-inflammatory, anti-atherosclerotic, and vasorelaxant effects [8-11].
As a part of our drug discovery program on antimalarial agents from Indian medicinal plants, we isolated large quantities of anthraquinone i.e. emodin (1) from the roots of Rheum emodi and planned to carry out chemical transformation to improve its therapeutic application. Chemical transformation of bioactive compounds of medicinal herbs is one of the most common approaches in drug discovery to improve the therapeutic properties. Towards this goal, we have synthesized a library of novel emodin derivatives 2-24 and their antimalarial activity was evaluated
General chemistry
IR spectra were recorded on perkin-Elmer RX-1 spectrometer. Using either KBr pellets (or) in neat. 1H-NMR,13C-NMR, DEPT-90 and DEPT-135 spectra were run on Bruker Advance DPX 300 MHz and 200 MHz in CDCl3.Chemical shifts are reported as values in ppm relative to CHCl3 (7.26) in CDCl3 and TMS was used as internal standard.ESI mass spectra were recorded on JEOL SX 102/DA-6000. Chromatography was executed with silica gel (60-120 mesh) using mixtures of chloroform, ethyl acetate and hexane as eluants
Background of plant
Rheum emodi wall (Family: Polygonaceae, commonly known as revand-chini and English name rhubarb) is a stout herb, distributed in the alpine and sub-alpine zones of the Himalayas. The roots of this species are used widely in ayurvedic medicine. Roots of the Indian rhubarb is darker, inferior in aroma, and is a well-known stomachic, bitter, cathartic and used all over the world.
Collection of medicinal plant
Rheum emodi wall (Bark) was purchased from the local market of Lucknow, U.P, India and the authentification was done by Botany Division of Central Drug Research Institute, Lucknow.
Extraction
Powdered Rheum emodi wall (Bark) (3 kg) were placed in glass percolator with 95% ethanol (10 lit) and allowed to stand for 24hr at room temperature. The percolate was collected and these processes were repeated for four times. The combined percolate was evaporated under reduced pressure at 50°C to afford ethanol extract. The weight of extract was found to be 200 g.
Isolation and purification of Emodin
The alcoholic extract was (200 g) chromatographed on a column of silica gel (60-120 mesh), eluted with hexane and chloroform (70:30); recrystallization from methanol afford emodin (3g). The compound visualization was obtained under UV light, also shown orange spot by spraying with 10% sulphuric acid in methanol.
1, 3, 8-trihydroxy-6-methylanthracene-9-10-dione (1)
IR (KBr) 3613, 2925, 1625, 1461, 1373, 1277, 1029, 767, 672 cm-1 ; 1H NMR (DMSO-d6, 300 MHz) δ 12.03 (s, OH), 11.95 (s, OH), 7.41 (s, 1H), 7.10 (s, 1H), 7.06 (s, 1H), 6.55 (s, 1H), 2.38 (s, 3H); 13 NMR(DMSO-d6, 75 MHz) δ 189.40, 180.92, 165.51, 164.35, 161.30, 148.04, 134.77, 132.47, 123.91, 120.25, 113.04, 108.09, 107.75, 21.45; MS (ESI) m/z 270.
Preparation of emodin derivatives
General method for O-alkylation (Method A): A stirred solution of compound 1 (100 mg, 0.00037 moles) in DMF (5 mL) at room temperature was treated with respective alkyl halide (RX) (0.00033 moles) and K2CO3 (0.00092 moles). The reaction mixture was stirred at 60-70°C for 4 hr. It was then extracted with ethyl acetate (3 × 25 mL), the organic layer was washed with water, dried over anhydrous Na2SO4 and evaporated under reduced pressure. Then the crude product was chromatographed on silica gel to afford the desired compound.
Ethyl 2 - (4, 5-dihydroxy-7-methyl-9, 10-dioxo-9, 10 dihydroanthracen- 2-yloxy)-2-methylpropanoate (2): IR (KBr) 3688, 3400, 2374, 1722, 1625, 1468, 1382, 1283, 1217, 1133, 766, 671 cm-1; 1H NMR (CDCl3, 300MHz) δ 12.22 (s, OH), 12.10 (s, OH), 8.02 (s, 1H), 7.62 (s, 1H), 7.09 (s, 1H), 6.56 (s, 1H), 4.31-4.24 (q, 2H), 2.45 (s, 3H), 1.71 (s, 6H), 1.42 (t, 3H); ESI - MS: Cacld for C21H20O7 [M+H]+: 384, Found 384; Yield: 65%
3-(2-(diethyl amino) ethoxy)-1, 8-dihydroxy-6-methylanthracene- 9, 10-dione (3): IR (KBr) 3869, 3413, 1638,1333, 1216, 1028, 925, 764, 672cm-1; 1H NMR (CDCl3, 300MHz,) δ 12.27 (s, OH), 12.09 (s, OH), 7.61 (s, 1H), 7.35 (s, 1H), 7.07 (s, 1H), 6.68 (s, 1H), 4.25 (t, 2H), 2.86 (t, 2H), 2.62 (s, 4H), 2.45 (s, 3H), 1.40 (t, 6H); ESI - MS: Cacld for C21H23NO5 [M+H]+: 370, Found 370; Yield: 70%.
1,8-dihydroxy-3-methyl-6-(2-(pyrrolidin-1-yl)ethoxy) anthracene-9, 10-dione (4): IR (KBr) 3438, 2341, 1720, 1630, 1463, 1385, 1285, 1218, 1130, 1074, 765, 668 cm-1; 1H NMR (CDCl3, 300MHz,) δ 7.59 (s, 1H), 7.32 (s, 1H), 7.05 (s, 1H), 6.66 (s, 1H), 4.29 (t, 2H), 2.95 (t, 2H), 2.66 (t, 4H), 2.43 (s, 3H), 2.04 (t, 4H); ESI - MS: Cacld for C21H21NO5 [M+H]+: 368, Found 368; Yield: 68%.
1, 8-dihydroxy-3-methyl-6-(2-(piperidin-1-yl) ethoxy) anthracene- 9, 10-dione (5): IR (KBr) 3285, 2923, 1725, 1623, 1461, 1376, 1218, 909, 764, 669 cm-1; 1H NMR (CDCl3, 300 MHz,) δ 7.61 (s, 1H), 7.35 (s, 1H), 7.08 (s, 1H), 6.68 (s, 1H), 4.31 (t, 2H), 2.96 (t, 2H), 2.68 (t, 4H), 2.45 (s, 3H), 1.43 (t, 6H); ESI - MS: Cacld for C22H23NO5 [M+H]+: 383, Found 383; Yield: 68%.
1, 8-dihydroxy-3-methyl-6-(2-morpholinoethoxy) anthracene-9, 10-dione (6): IR (KBr) 3677, 3646, 3018, 1626, 1462, 1217, 920, 767, 670 cm-1; 1H NMR (CDCl3, 300MHz,) δ 7.60 (s, 1H), 7.34 (s, 1H), 7.06 (s, 1H), 6.67 (s, 1H), 4.24 (t, 2H), 3.74 (t, 4H), 2.85 (t, 3H), 2.62 (t, 4H), 2.44 (s, 3H); ESI - MS: Cacld for C21H21NO6 [M+H]+: 384, Found 384; Yield: 70%.
1, 8-dihydroxy-3-methyl-6-(nonyloxy) anthracene-9, 10-dione (7): IR (KBr) 3365, 2359, 1628, 1461, 1217, 1020, 769, 671 cm-1; 1H NMR (CDCl3, 300 MHz) δ 12.27 (s, OH), 12.10 (s, OH), 7.59 (s, 1H), 7.32 (s, 1H), 7.05 (s, 1H), 6.64 (s, 1H), 4.07 (t, 2H), 2.43 (s, 3H), 1.82 - 1.78 (m, 2H), 1.58 (s, 2H), 1.45 (s, 2H), 1.25 (s, 6H), 0.85 (t, 3H); MS (ESI) m/z 382; Yield:66%.
1, 8-dihydroxy-3-methyl-6-(undecyoxy) anthracene-9, 10-dione (8): IR (KBr) 3761, 3414, 2952, 1624, 1318, 1216, 766, 671 cm-1; 1H NMR (CDCl3, 300 MHz,) δ 12.49 (s, OH), 12.32 (s, OH), 7.81 (s, 1H), 7.54 (s, 1H), 7.46 (s, 1H), 6.84 (s, 1H), 4.27 (t, 2H), 2.64 (s, 3H), 2.02 - 1.99 (m, 2H), 1.76 (s, 2H), 1.66 (s, 2H), 1.45 (s, 16H), 1.07 (t, 3H) ); 13C NMR(CDCl3, 75 MHz) δ 191.06, 183.04, 167.18, 166.19, 163.45, 149.31, 140.27, 136.16, 134.23, 125.44, 122.21, 115.06, 114.70, 111.00, 109.72, 108.10, 70.04, 34.02, 32.93, 32.63, 30.70, 30.50, 30.53, 30.36, 30.29, 30.16, 29.96, 29.89, 26.89, 23.69, 23.13, 15.11; MS (ESI) m/z 438; Yield: 65%.
3-(dodecyloxy)-1, 8-dihydroxy-6-methyl anthracene-9, 10-dione (9): IR (KBr) 3429, 1627, 1473, 1386, 1303, 1264, 1218, 1032, 769, 670 cm-1; 1H NMR (CDCl3, 300 MHz,) δ 12.30 (s, OH), 12.13 (s, OH), 7.62 (s, 1H), 7.35 (s, 1H), 7.07 (s, 1H), 6.85 (s, 1H), 4.08 (t, 2H), 2.44 (s, 3H), 2.05 (s, 2H), 1.85 - 1.80(m, 2H), 1.57 (s, 2H), 1.45 - 1.41 (m, 4H), 1.29 (s, 2OH), 0.86 (t, 3H); 13 NMR(CDCl3, 75 MHz) δ 190.65, 182.01, 166.16, 165.17, 162.44, 148.31, 136.12, 133.19, 124.43, 121.21, 106.71, 107.08, 69.03, 31.92, 31.62, 29.66, 29.36, 28.88, 25.88, 22.69, 22.13, 14.12; MS (ESI) m/z 452; Yield: 65%.
1,8-dihydroxy-3-methyl-6-(pentadecyloxy)anthracene-9, 10-dione (10): IR (KBr) 3490, 2924, 1627, 1452, 1217, 769, 670 cm-1; 1H NMR (CDCl3, 300 MHz,) δ 7.61 (s, 1H), 7.34 (s, 1H), 7.06 (s, 1H), 6.65 (s, 1H), 4.07 (t, 2H), 2.44 (s, 3H), 2.02 (t, 2H), 1.84 - 1.79 (m, 2H), 1.56 (s, 2H), 1.25 (s, 24H), 0.87 (t, 3H); 13 NMR(CDCl3, 75 MHz) δ 190.69, 182.05, 166.19, 166.20, 162.47, 148.32, 139.27, 135.17, 133.24, 124.45, 121.22, 114.06, 113.70, 110.03, 108.72, 107.10, 69.04, 33.83, 31.93, 31.63, 29.70, 29.54, 29.37, 29.30, 29.16, 28.96, 28.89, 25.89, 22.70, 22.13, 14.12; MS (ESI) m/z 494; Yield: 62%.
1,8-dihydroxy-3-methyl-6-(octadecyloxy)anthracene-9, 10-dione (11): IR (KBr) 3407, 2952, 1631, 1363, 1217, 1012, 769, 670 cm-1; 1H NMR (CDCl3, 300 MHz,) δ 12.29 (s, OH), 12.12 (s, OH), 7.61 (s, 1H), 7.34 (s, 1H), 7.07 (s, 1H), 6.66 (s, 1H), 4.09 (t, 2H), 2.45 (s, 3H), 1.86 - 1.82 (m, 2H), 1.60 (s, 2H), 1.47 - 1.43 (m, 2H), 1.27 (s, 28H), 0.87 (t, 3H) ; MS (ESI) m/z 522; Yield: 65%.
1, 8-dihydroxy-3-methyl-6-(icosyloxy) anthracene-9, 10-dione (12): IR (KBr) 3289, 2368, 1658, 1584, 1458, 1218, 769, 670 cm-1;1H NMR (CDCl3, 300 MHz,) δ 12.31 (s, OH), 12.15 (s, OH), 7.64 (s, 1H), 7.37 (s, 1H), 7.09 (s, 1H), 6.68 (s, 1H), 4.10 (t, 2H), 2.46 (s, 3H), 2.05 (t, 2H), 1.87 - 1.82 (m, 2H), 1.59 (s, 2H), 1.34 (s, 31H), 0.88 (t, 3H) ); 13 NMR(CDCl3, 75 MHz) δ 190.11, 181.47, 165.61, 164.62, 161.89, 147.74, 138.69, 134.59, 132.66, 123.87, 120.54, 113.48, 113.12, 109.45, 108.14, 106.52, 68.45, 32.25, 31.35, 31.05, 29.12, 29.0, 28.79, 28.72, 28.59, 28.38, 28.31, 25.31, 22.12, 21.55, 13.54; MS (ESI) m/z 550; Yield: 63%.
3-(docosyloxy)-1, 8-dihydroxy-6-methyl anthracene-9, 10-dione (13): IR(KBr) 3388, 2362, 1624, 1365, 1217, 767, 672 cm-1;1H NMR (CDCl3, 300 MHz,) δ 12.29 (s, OH), 12.12 (s, OH), 7.61 (s, 1H), 7.34 (s, 1H), 7.06 (s, 1H), 6.65 (s, 1H), 4.08 (t, 2H), 2.44 (s, 3H), 2.05 (t, 2H), 1.82 (s, 2H), 1.25 (s, 34H), 0.88 (t, 3H); MS (ESI) m/z 578; Yield: 64%.
(E)-3-(3, 7-dimethylocta-2, 6-dienyloxy) 1, 8-dihydroxy-6- methyl anthracene-9, 10-Dione (14): IR (KBr) 3349, 2367, 1718, 1624, 1364, 1218, 770, 650 cm-1; 1H NMR (CDCl3, 300 MHz,) δ 1H 12.32 (s, OH), 12.16 (s, OH), 7.64 (s, 1H), 7.28 (s, 1H), 7.10 (s, 1H), 6.70 (s, 1H), 5.51 (t, 1H), 5.11 (s, 1H), 4.71 (t, 2H), 2.47 (s, 3H), 2.14 (s, 4H), 1.80 (s, 6H), 1.73 (s, 3H); MS (ESI) m/z 406; Yield: 62%.
1, 8-dihydroxy-3-methyl-6-(prop-2-ynyloxy) anthracene-9, 10-dione (15): IR (KBr) 3282, 2922, 2125, 2357, 1717, 1631, 1390, 1218, 1078, 766, 680 cm-1; 1H NMR (CDCl3, 300 MHz) δ 12.12 (s, OH), 12.01 (s, OH), 7.80 (s, 1H), 7.66 (s, 1H), 7.64 (s, 1H), 7.096 (s, 1H), 4.03 (s, 2H), 2.85 (s, 1H), 2.46 (s, 3H); MS (ESI) m/z 308; Yield: 60%.
General method for C-alkylation (Method B)
A stirred solution of amine (0.0022 moles) cooled at 0°C was slowly treated with formalin (0.00148 moles), glacial acetic acid (0.0192 moles) and compound 1 (100 mg, 0.00037 moles). The whole reaction mixture was brought to room temperature and stirred for 4 hr, after pH was adjusted to 8 by addition of 20% aq. NaOH. It was then extracted with ethyl acetate (3 × 25 mL), the organic layer was washed with water, dried over anhydrous Na2SO4 and evaporated under reduced pressure. Then the crude product was chromatographed on silica gel to afford the desired compound.
1,3,8-trihydroxy-6-methyl-2-(pyrrolidin-1-ylmethyl) anthracene-9, 10-dione (16): IR (KBr) 3556, 3023, 2925, 2357, 1625, 1379, 1278, 1104, 767, 671 cm-1;1H NMR (DMSO-d6,300MHz) δ 7.40 (s, 1H), 6.85 (s, 1H), 6.78 (s, 1H), 4.14 (s, 2H), 3.21 (t, 4H), 2.36 (s, 3H), 1.96 (t, 4H); ESI - MS: Cacld for C20H19NO5 [M+H]+: 354, Found 354; Yield: 80%.
1,3,8-trihydroxy-6-methyl-2-(piperidin-1-ylmethyl) anthracene-9, 10-dione (17): IR(KBr) 3401, 2927, 2372, 1628, 1535, 1459, 1369, 1222, 1112, 768 cm-1;1H NMR (DMSO-d6,300MHz) δ 7.39 (s, 1H), 6.90 (s, 2H), 3.90 (s, 2H), 2.74 (t, 4H), 2.33 (s, 3H), 1.68 (t, 4H), 1.52 (t, 2H); ESI - MS: Cacld for C21H21NO5 [M+H]+: 369, Found 369; Yield: 78%.
1,3,8-trihydroxy-6-methyl-2-(morpholinomethyl) anthracene-9, 10-dione (18): IR (KBr) 3565, 3021, 2929, 2357, 1720, 1618, 1461, 1377, 1283, 1217, 1122, 765, 671 cm-1; 1H NMR (CDCl3 & DMSO-d6, 300MHz) δ 7.43 (s, 1H), 6.86 (s, 1H), 6.70 (s, 1H), 3.65 (t, 2H), 3.49 (t, 4H), 2.54 (t, 2H), 2.27 (t, 2H), 2.17 (s, 3H); ESI - MS: Cacld for C21H23NO5 [M+H]+: 370, Found 370; ESI - MS: Cacld for C20H19NO6 [M+H]+: 370, Found 370; Yield: 80%.
1,3,8-trihydroxy-6-methyl-2-(piperazin-1-ylmethyl) anthracene-9, 10-dione (19): IR (KBr) 3463, 3020, 2929, 2356, 1720, 1649, 1458, 1380, 1284, 1217, 1123, 765, 670 cm-1; 1H NMR (CDCl3, 300MHz) δ 12.78 (s, OH), 12.05 (s, OH), 7.66 (s, 1H), 7.02 (s, 1H), 6.86 (s, 1H), 3.97 (s, 2H), 3.03 (t, 4H), 2.52 (t, 2H), 2.48 (t, 2H), 2.32 (s, 3H), 2.10(s, 1H); ESI - MS: Cacld for C20H20N2O5 [M+H]+: 369, Found 369; Yield: 75%.
1, 3, 8-trihydroxy-6-methyl-2-((4-methylpiperazin-1-yl) methyl) anthracene-9, 10-dione (20): IR (KBr) 3484, 3021, 2403, 1678, 1526, 1427, 1216, 929,762, 672 cm-1; 1H NMR (CDCl3 & DMSO-d6, 300MHz ) δ 7.84 (s, 1H), 7.69 (s, 1H), 7.20 (s, 1H), 4.11 (s, 2H), 2.92 (t, 4H), 2.76 (t, 4H), 2.61 (s, 3H), 2.49 (s, 3H) ); 13 NMR(CDCl3 & DMSO, 75 MHz) 190.62, 189.40, 168.45, 163.03, 162.67, 148.47, 134.58, 133.71, 124.73, 121.31, 113.25, 110.66, 108.59, 54.88, 53.96, 52.92, 46.22, 22.58; ESI - MS: Cacld for C21H22N2O5 [M+H]+: 383, Found 383; Yield: 78%.
2-((4-(4-fluorophenyl) piperazin-1-yl) methyl)-1, 3, 8-trihydroxy-6-methylanthracene-9, 10-dione (21): IR (KBr) 3557, 2931, 2837, 2355, 1650, 1510, 1455, 1367, 1222, 1018, 926, 766, 669 cm-1 ; 1H NMR (CDCl3, 300MHz,) δ 12.66 (s, OH),12.05 (s, OH), 7.52 (s, 1H), 7.15 (s, 1H), 6.98 (s, 1H), 6.89 (m,4H), 3.93 (s, 2H), 3.13 (t, 4H), 2.77 (t, 4H), 2.36 (s, 3H); ESI - MS: Cacld for C26H23FN2O5 [M+H]+: 463, Found 463; Yield: 80%.
General method for esterification and acid formation (Method C): A stirred solution of compound 1 (100mg, 0.00037 moles) in pyridine (2 mL) and acetic anhydride (0.00185 moles) at 60-70°C for 4 hr. The reaction mixture was put into cold water for crystallization, then filtered and dried. To the resultant crude acetate was gradually added 10 ml of acetic anhydride and glacial acetic acid mixture (1:1) and CrO3 at 45°C and stirred for 10 hr at 70°C. Acetic anhydride and glacial acetic acid mixture was removed by vacuum. It was then extracted with ethyl acetate (3 × 25 mL), the organic layer was washed with water, dried over anhydrous Na2SO4 and evaporated under reduced pressure. Then the crude product was chromatographed on silica gel to afford the desired compound.
(E)-4,5-dihydroxy-7-methyl-9,10-dioxo-9, 10-dihydroanthracen- 2-yl 2-methylbut-2-enoate (22): IR (KBr) 3408, 3023, 2929, 2370, 1731, 1639, 1464, 1373, 1217, 1119, 926, 764, 672 cm-1; 1H NMR (CDCl3, 300MHz) 12.57 (s, OH), 12.23 (s, OH), 8.02 (s, 1H), 8.01 (s, 1H), 7.63 (s, 1H), 7.33-7.19 (q, 1H), 7.11 (s, 1H), 2.46 (s, 3H), 2.05 (s, 3H), 1.98 (d, 3H); ESI - MS: Cacld for C20H16O6 [M+H]+: 352, Found 352; Yield: 65%.
6-methyl-9, 10-dioxo-9,10-dihydroanthracene-1,3,8-triyl triacetate (23): IR (KBr) 3407, 2927, 1761, 1668, 1605, 1457, 1324, 1206, 1029, 908, 766, 674 cm-1; 1H NMR (CDCl3, 300MHz) 8.01 (s, 1H), 7.95 (s, 1H), 7.23 (s, 1H), 7.22 (s, 1H), 2.50 (s, 3H), 2.43 (s, 6H), 2.35 (s, 3H); ESI - MS: Cacld for C21H16O8 [M+H]+: 397, Found 397; Yield: 98%.
4,5,7-triacetoxy-9,10-dioxo-9,10-dihydroanthracene-2- carboxylic acid (24): IR (KBr) 3416, 3024, 2927, 2367, 1770, 1648, 1460, 1372, 1215, 1029, 923, 761, 672 cm-1; 1H NMR (DMSO-d6,300MHz) 8.12 (s, 1H), 7.95 (s, 1H), 7.93 (s, 1H), 7.63 (s, 1H), 2.39 (s, 9H); 13 NMR (DMSO-d6,75 MHz) 180.14, 179.37, 168.40, 166.29, 165.02, 154.69, 150.99, 149.81, 136.56, 135.26, 134.43, 132.51, 124.98, 123.96, 123.08, 121.93, 120.39, 20.88; ESI - MS: Cacld for C21H14O10 [M+H]+: 425, Found 425; Yield: 80%.
Materials
RPMI-1640 medium, sodium bicarbonate, glucose, hypoxanthine, resazurin, chloroquine diphosphate and MEM medium were purchased from Sigma (St. Louis, MO, USA). Albumax II was procured from Gibco BRL (Grand Island, NY, USA). Giemsa stain was purchased from Merck (USA). SYBR Green l nucleic acid gel stain was purchased from Invitrogen molecular probe (Carlsbad, USA).
In-vitro cultivation of P. falciparum
The chloroquine sensitive (Pf3D7) and chloroquine resistant (PfK1) strains of P. falciparum were grown in continuous culture according to Trager and Jensen [12]. Parasites were cultured in RPMI-1640 (HEPES modified) medium (Sigma) supplemented with 0.5% AlbuMaxII, 0.2% glucose, 0.2% NaHCO3 and 15 μM hypoxanthine. Parasite growth rate and stage was determined by the examination of Giemsa’s stained thin smears of the RBCs.
Antimalarial activity of compounds
In-vitro assessment of antimalarial activity of compounds towards P. falciparum was performed by the determination of fifty percent inhibitory concentration (IC50) according to the method of Johnson [13] with some modifications. Compounds were tested in range between 20 μM to 0.31 μM. To determine IC50 initially 20 μM concentration were used and two fold serially diluted till 0.31 μM and chloroquine were prepared in 96 well plates and then 50 μl asynchronous culture of infected erythrocytes with 1-1.5% parasitaemia and 2-3% haematocrit was added to each well (100 μl-final volume). Eight wells were treated as positive control (without drug) and 4 wells as negative controls (without parasite and drug). These plates were incubated in CO2 incubator maintained at 37°C for 72 h. After 72 h, 100μl lytic buffer containing SYBR Green 1X final concentration was added to each well and incubated for 1-2h at room temperature in dark. Plates were read under fluorescence reader (Synergy HT BioTek) at Ex. 485nm, Em. 535nm. IC50 was determined on the basis of DNA content of the parasite by using MS-Excel template.
Cytotoxicity assay
Cytotoxic level of active compounds would be assessed according to protocol defined by O’Brien [14], with some modifications. The monkey kidney cell line will be maintained in vitro in MEM medium supplied with 15% Fetal Bovine Serum (FBS) and 5% CO2 at 37°C. An appropriate serial drug dilution was prepared in culture plates and the cells were exposed to these concentrations of particular compounds for two days, 10% of cell viability marker resazurin was added and read under fluorescent reader at excitation wavelength 530 ± 25nm and emission at 590 ± 25 nm for calculation of the median cytotoxic concentration (CC50). The selective index (SI) would be calculated by using the formula-

Statistical analysis
Fifty percent inhibitory concentration (IC50) of tested compounds were obtained by transferring the data into a graphic program (e.g. Excel) and expressed as percentage of the untreated controls and then evaluated by Logit regression analysis using pre-programmed Excel spreadsheet obtained from MMV group at Swiss Tropical Institute, Basel, Switzerland [15].
Chemistry
In order to prepare O-alkylated derivatives (2-6), (7-15) hydroxyl group at meta position of emodin (1) was alkylated with various amine side chains (Figure 1) and long chain alkyl halides (Figure 1) under basic conditions using K2CO3 and DMF as a solvent.
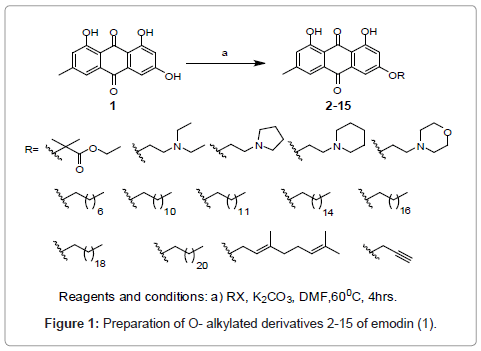
Figure 1: Preparation of O- alkylated derivatives 2-15 of emodin (1).
To study the substituent effect on anthraquinone nucleus C-alkylated derivates 16-21 were prepared using Mannich reaction protocol [16-20] (Figure 2).
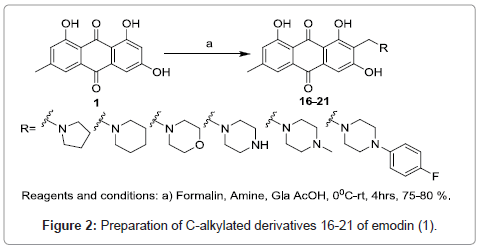
Figure 2: Preparation of C-alkylated derivatives 16-21 of emodin (1).
“Since the emodin (1) contain free hydroxyl group we planned to” prepare few esters to study their antimalarial activity. To prepare ester derivative 22 and 23, emodin (1) was reacted with tiglic acid in presence of triethylamine at 0°C and acetic anhydride in presence of pyridine respectively. To transform the methyl group in to acid functionality emodin triacetate (23) was oxidized with chromium trioxide in presence of glacial acetic acid and acetic anhydride to provide acid (24).
Biological evaluation
In vitro antimalarial activity: All the compounds were screened against chloroquine sensitive (Pf3D7) and chloroquine resistant (PfK1) strain of P. falciparum. The parent compound, emodin (1) has an IC50 of 16.2 and 37 μM against Pf3D7 and PfK1 strain respectively (Table 1). Except 2, 3, 7-11, 13, 14 & 24 all other derivatives exhibited improved in-vitro antimalarial activity against Pf3D7 and PfK1 strains with better therapeutic index (Table 1). Derivatives 4 (IC50=12.6 μM), 6 (IC50=10.6 μM), 15 (IC50=11.6 μM), 16 (IC50=10.1 μM), 19 (IC50=10.05 μM), 22 (IC50=13.55 μM) shows moderate activity against Pf3D7. Derivatives 5 (IC50=12.1 μM), 23 (IC50=19.09 μM) shows moderate activity against PfK1. Derivatives 5 (IC50=5.7 μM), 12 (IC50=2.1 μM) have IC50 below 10 μM against Pf3D7. Derivatives 16 (IC50=6.26 μM), 19 (IC50=9.32 μM), 21 (IC50=8.29 μM) have IC50 below 10 μM against PfK1.Where as derivatives 17 (IC50=2.74 μM, 2.28 μM), 18 (IC50=5.36 μM, 2.49 μM), 20 (IC50=3.58 μM, 2.48 μM) also have IC50 below 10 μM against Pf3D7 and PfK1 strain. Our structure activity relationships indicated that C-alkylation of emodin (1) improves the activity in both strains, where as O-alkylation only improves the activity in Pf3D7 strain. It is noteworthy to mention here that Mannich base derivatives 17, 18 and 20 exhibited comparable in-vitro antimalarial activity with the marketed drug chloroquine (IC50= 1.12) with good therapeutic index against CQ resistant strain (Table 2, Figure 3 and 4).
| Comp. No | Chemical Structure | IC50 | CC50 | |
|---|---|---|---|---|
| Pf3D7 (CQS) |
PfK1 (CQR) |
VERO cell line | ||
| µM | µM | µM | ||
| 1 | 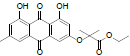 |
16.2 | 37 | nd |
| 2 | 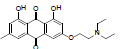 |
16.2 | >52 | nd |
| 3 | 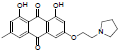 |
22.4 | 48.8 | nd |
| 4 | 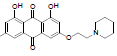 |
12.6 | 30.6 | nd |
| 5 | 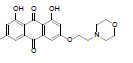 |
5.7 | 12.1 | 69.8 |
| 6 | 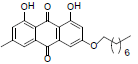 |
10.6 | 18.41 | nd |
| 7 | 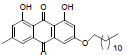 |
36.5 | >52.35 | nd |
| 8 | 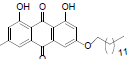 |
>45.6 | >45.6 | nd |
| 9 | 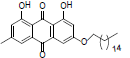 |
>44.24 | >44.24 | nd |
| 10 | 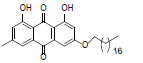 |
>40.4 | >40.4 | nd |
| 11 | 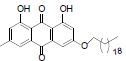 |
>38.31 | >38.31 | nd |
| 12 | 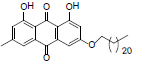 |
2.1 | >36.36 | 909 |
| 13 | 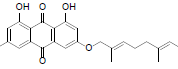 |
>34.6 | >34.6 | nd |
| 14 | 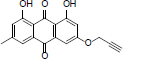 |
>49.26 | >49.26 | nd |
| 15 | 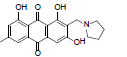 |
11.6 | >64.93 | 1623 |
| 16 |  |
10.1 | 6.26 | nd |
| 17 | 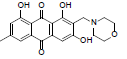 |
2.74 | 2.28 | >1358 |
| 18 |  |
5.36 | 2.49 | >1355 |
| 19 | 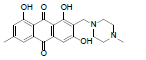 |
10.05 | 9.32 | nd |
| 20 | 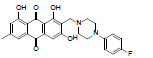 |
3.58 | 2.48 | 782.7 |
| 21 |  |
18.74 | 8.29 | nd |
| 22 | 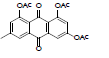 |
13.55 | 52.89 | nd |
| 23 | 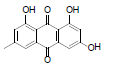 |
25.93 | 19.09 | nd |
| 24 | 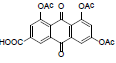 |
30.04 | 34.50 | nd |
| Chloroquine (Standard antimalarial) |
0.014 | 1.12 | 226.7 | |
Table 1: Chemical structures and in vitro antimalarial activity (IC50 in μM) of emodin (1) and its derivatives (2–24).
| Compound No. |
IC50 (Pf3D) (µM) |
IC50 (Pfk1) (µM) |
SI (Pf3D7) (µM) |
SI (Pfk1) (µM) |
|---|---|---|---|---|
| 17 | 2.74 | 2.28 | 495.6 | 595.6 |
| 18 | 5.36 | 2.49 | >252 | >544 |
| 20 | 3.58 | 2.48 | 218.6 | 315 |
| Chloroquine | 0.014 | 1.12 | 16192.8 | 201.7 |
Table 2: Antimalarial profile and safety index of active compounds against CQS (Pf3D7) and CQR (PfK1) strains of P.falciparum.
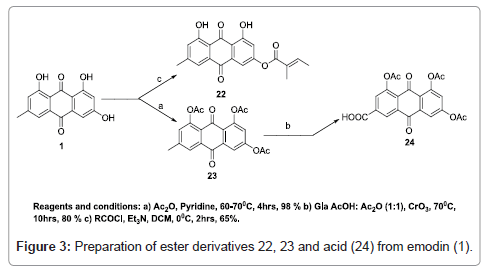
Figure 3: Preparation of ester derivatives 22, 23 and acid (24) from emodin (1).
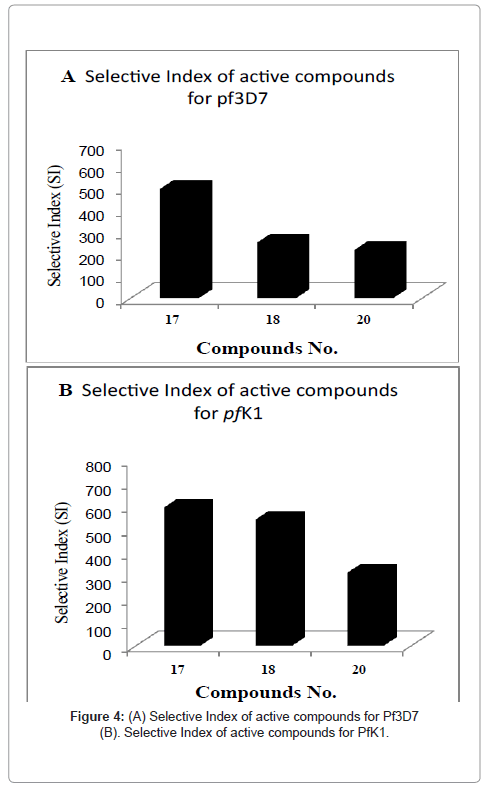
Figure 4: (A) Selective Index of active compounds for Pf3D7
(B). Selective Index of active compounds for PfK1.
Cytotoxicity assay and selective index (SI)
In order to characterize the Pf3D7 and PfK1 strain basis of antimalarial effects of selected derivatives 5, 12, 15, 17, 18 and 20 we investigated the cytotoxic level of these active compounds. Derivatives 5 (CC50=69.8 μM), 12 (CC50=909 μM), 15 (CC50=1623 μM), 17 (CC50= >1358 μM), 18 (CC50= >1355 μM), 20 (CC50=782.7 μM) against VERO cell line. These compounds did not show any significant cytotoxicity against vero cell line (Table 4).
In conclusion, we have isolated larger quantities of emodin (1) from the roots of Rheum emodi and a library of novel emodin O-alkylated derivatives 2-6, 7-15, C-alkylated Mannich derivatives 16-21, acyl derivatives 22-23, and acid derivative 24 (Figure 3) from 1 were synthesized and evaluated their in-vitro antimalarial activity against chloroquine sensitive strain and chloroquine resistant strain. Among these 24 derivatives, C-alkyl Mannich bases 17, 18 and 20 showed potent antimalarial activity against chloroquine resistant strain PfK1with an IC50 of 2.28, 2.49, 2.48 μM respectively, which is comparable to marketed drug chloroquine (IC50 of 1.12). Further work is in progress in our laboratory to prepare more Mannich bases of emodin to develop a potent lead for inhibition of chloroquine resistant malaria.
Authors are thankful to Director, CSIR-CDRI for constant encouragement for the program on natural products of biological importance. We also thank the ICMR, New Delhi for providing research grant and fellowship. SAIF, CDRI for spectral data. Financial support from SPlenDID is acknowledged.
